Síndrome de Dandy-Walker
Origem: Wikipédia, a enciclopédia livre.
A síndrome de Dandy-Walker é uma malformação congénita que afecta o cerebelo e os fluidos que o rodeiam. As características principais desta síndrome são o alargamento do quarto ventrículo, a ausência completa ou parcial da área entre os dois hemisférios cerebelares e a formação de cistos na base interna do crânio.Malformação de Dandy-Walker (DWM) é a associação dos sinais e sintomas: hidrocefalia, parcial ou completa, ausência do vermis cerebelar, cisto da fossa posterior e contíguo com o quarto ventrículo.
A incidência desta síndrome é de cerca de um por 100.000 nascidos vivos, sendo 1,24 para o sexo masculino e 0,78 para o sexo feminino.
Esta síndrome é responsável por 3,5% dos casos de hidrocefalia infantil.
Os pacientes apresentam nos primeiros anos de vida com hidrocefalia associada com abaulamento occipício. Outros sinais são fossa posterior, como sinais palsies dos nervos cranianos, apresentam ainda ataxia e nistagmo.
Radiologicamente os pacientes apresentam elevadas logomarca dos seios transverso do desbaste e abaulamento dos ossos da fossa posterior.
A anomalia é etiologicamente heterogênea e uma revisão da literatura demonstrou que, de acordo com a história familiar e anomalias associadas, a Dandy Walker malformação pode ser autossômica recessiva, em uma série de malformações de múltiplas síndromes, como por exemplo, síndrome de Meckel ou Síndrome de Joubert. Pode ainda ser de herança autossômica recessivo em caso de erros inatos do metabolismo, como por exemplo, na síndrome CDG e autossômica dominante como, por exemplo, na síndrome G e ligada ao cromossomo X recessivo, como por exemplo, na síndrome de Aicardi.
Nos pacientes com esta síndrome parece haver uma maior freqüência de associação com cardiopatias congênitas, fissura lábio palatal e defeitos do tubo neural.
O diagnóstico pré-natal é possível por que a ultra-sonografia pode mostrar cisto da fossa posterior e agenesia vermis cerebelar.
Veja mais sobre hidrocefalia infantil, criança com hidrocefalia, síndrome de Dandy-Walker, síndromes e doenças raras, malformações cerebelar, acesse estas categorias no site ou clique nos links desta página.
************************************************************************
Anencefalia
Origem: Wikipédia, a enciclopédia livre.
| Anencefalia |
||
| Classificação e recursos externos | ||
 |
||
|---|---|---|
| Uma visão frontal de um feto com anencefalia | ||
| CID-10 | Q00.0 | |
| CID-9 | 740.0 | |
| OMIM | 206500 | |
| DiseasesDB | 705 | |
| MeSH | C10.500.680.196 | |
Ao contrário do que o termo possa sugerir, a anencefalia não caracteriza casos de ausência total do encéfalo, mas situações em que se observam graus variados de danos encefálicos. A dificuldade de uma definição exata do termo "baseia-se sobre o fato de que a anencefalia não é uma má-formação do tipo 'tudo ou nada', ou seja, não está ausente ou presente, mas trata-se de uma má-formação que passa, sem solução de continuidade, de quadros menos graves a quadros de indubitável anencefalia. Uma classificação rigorosa é, portanto quase que impossível".[1]
Na prática, a palavra "anencefalia" geralmente é utilizada para caracterizar uma má-formação fetal do cérebro. Nestes casos, o bebê pode apresentar algumas partes do tronco cerebral funcionando, garantindo algumas funções vitais do organismo.[2]
Trata-se de patologia letal. Bebês com anencefalia possuem expectativa de vida muito curta, embora não se possa estabelecer com precisão o tempo de vida que terão fora do útero. A anomalia pode ser diagnosticada, com certa precisão, a partir das 12 semanas de gestação, através de um exame de ultra-sonografia, quando já é possível a visualização do segmento cefálico fetal. [carece de fontes]
O risco de incidência aumenta 5% a cada gravidez subsequente. Inclusive, mães diabéticas têm seis vezes mais probabilidade de gerar filhos com este problema. Há, também, maior incidência de casos de anencefalia em mães muito jovens ou nas de idade avançada. Uma das formas de prevenção mais indicadas é a ingestão de ácido fólico antes e durante a gestação.[3]
A anencefalia é um dos três principais defeitos do tubo neural (DTN). Os outros são a encefalocele e a mielomeningocele. Os defeitos do tubo neural resultam de uma falha no fechamento do tubo neural que ocorre entre 25 e 27 dias após a concepção.
Nos Estados Unidos, a anencefalia ocorre em 1 a cada 10.000 nascimentos.[4] A malformação é mais comum em meninas[5], brancos e em mães nos extremos de idade. A taxa de anencefalia em nascidos vivos provavelmente subestima a real taxa de ocorrência da doença, pois diversos casos de aborto espontâneo são causados por fetos afetados que não chegam a receber o diagnóstico.
A prevalência ao nascimento diminuiu nos Estados Unidos após a suplementação obrigatória de alimentos com ácido fólico, que iniciou em janeiro de 1998.
Um recém-nascido com anencefalia geralmente é cego, surdo, inconsciente e incapaz de sentir dor. Embora alguns indivíduos com anencefalia possam nascer com um tronco encefálico, a falta de um cérebro funcionante descarta a possibilidade de vir a ter consciência e ações reflexas, como a respiração e respostas aos sons ou toques.
A anencefalia frequentemente pode ser diagnosticada no pré-natal através de um exame de ultrassom. O diagnóstico ultrassonográfico tem alta acurácia e é baseado na ausência do cérebro e da calota craniana. Outra característica que pode ser observada na ultrassonografia é a polidramnia, que ocorre em até 50% dos casos durante o 2º e 3º trimestres de gestação devido à menor deglutição do feto.
A dosagem de alfafetoproteína (AFP) sérica materna [6] e o ultrassom fetal[7] são úteis para rastreio de defeitos do tubo neural como espinha bífida ou anencefalia.
Às vezes a anencefalia não é diagnosticada, pois o feto acaba evoluindo para aborto espontâneo. Em outros, principalmente em mulheres que não têm acesso ao pré-natal, a doença é diagnosticada apenas durante o parto.
Prognóstico
Não existe cura ou tratamento padrão para a anencefalia e o prognóstico para estes pacientes é a morte. A maioria dos fetos não sobrevivem ao nascimento, o que corresponde a 55% dos casos não abortados. Quando a criança não é um natimorto (nasce sem vida), ela geralmente morre de parada cardiorrespiratória em poucas horas ou dias após o nascimento.[8][9]Entretanto, já existiram casos relatados de anencefalia que os pacientes sobreviveram até 2 anos após o nascimento.

O caso gerou divergências: alguns especialistas, baseados na deficiência de uma definição exata do termo "anencefalia", levantaram a hipótese de que a menina na verdade sofria de uma malformação do crânio (encefalocele), associada a um desenvolvimento reduzido do cérebro (microcefalia). Outros afirmam que o que houve, na verdade, foi uma forma "não clássica" de anencefalia, como avaliou a pediatra da menina, Márcia Beani Barcellos, profissional que mais acompanhou o caso. Segundo Márcia, a sobrevivência surpreendente de Marcela foi "um exemplo de que um diagnóstico não é nada definitivo".[11]
Interrupção da gravidez
A interrupção da gravidez, também conhecida como aborto terapêutico, é permitida em casos de anencefalia em diversos países.O Brasil autorizou em 2012 a realização do aborto terapêutico para fetos com anencefalia. Até então, grávidas com fetos com anencefalia precisavam de autorização judicial para realizar o aborto.
Segundo grupos contrários à manutenção da vida do feto com anencefalia, a interrupção da gravidez nestes casos diferiria do aborto por interromper o desenvolvimento de um feto que inevitavelmente morreria durante este processo, ou logo após o parto, enquanto o aborto interromperia o desenvolvimento de um bebê normal.[12] A interrupção da gravidez seria um processo semelhante, neste caso, a tirar a vida de uma pessoa em estado terminal, a qual sabe-se que inevitavelmente irá morrer, mais cedo ou mais tarde - no caso da anencefalia, provavelmente muito cedo.[13]. Essa visão é, entretanto, contestada por grupos contrários ao aborto, que alegam que toda vida tem valor, independente de seu tempo de duração.[12]
Um estudo realizado na Alemanha, onde o aborto terapêutico é permitido, demonstrou que as mães optam menos pela interrupção da gravidez em casos de anencefalia do que em casos de síndrome de Down (trissomia do 21) (5.642 abortos em 6.141 diagnósticos pré-natais de trissomia do 21 = 91,9%; 483 abortos de 628 casos de anencefalia = 76,9%; 358 abortos de 487 casos de espinha bífida = 73,5%).[14]
=================================================================================
Síndrome de Arnold-Chiari
Origem: Wikipédia, a enciclopédia livre.
A síndrome de Arnold-Chiari, ou má formação de Arnold-Chiari, consiste em uma mal formação rara e congênita do sistema nervoso central, localizada na fossa posterior da base cerebral. Esta malformação possui uma variabilidade de sinais e sintomas sendo que as principais consistem em alterações na estrutura do tronco cerebral e algumas vezes acompanhado de hidrocefalia.
A forma mais grave desta patologia consiste na herniação da porção mais
baixa do cerebelo e do tronco cerebral através do forame magno,
ocorrendo que algumas partes do cérebro alcançam o canal da medula espinhal comprimindo-a. O seu diagnóstico é confirmado pela Ressonância nuclear magnética e o tratamento consiste na descompressão do canal medular.O Chiari tipo 1 é uma mal formação do crânio que acontece na altura da junção entre o pescoço e a cabeça. Ocorre quando uma parte do encéfalo chamada de cerebelo, entra no canal vertebral (Figura). Esta deformidade está relacionada a um problema na circulação do líquido céfalo-raquidiano (líquor). Este líquido envolve todo o sistema nervoso central, no crânio e no canal vertebral e, quando há um distúrbio da sua circulação, pode ocasionar uma série de sinais e sintomas. A síndrome pode aparecer também em pessoas que não apresentem qualquer deformidade, como resultado de outras doenças, no entanto, a forma mais comum é a congênita e acomete principalmente as mulheres.
Os sintomas costumam aparecer na fase adulta entre a terceira e quarta décadas de vida e os mais comuns são: dor cervical, dor de cabeça intensa, fraqueza muscular, dormência ou alteração da sensibilidade nos membros e dificuldade de equilíbrio. Outros sintomas que podem surgir são: vertigem, distúrbios visuais, zumbidos, dificuldade para engolir, palpitação, apnéia do sono, diminuição das habilidades motoras finas e fadiga crônica. O exame neurológico realizado pelo especialista auxilia na determinação do diagnóstico, uma vez que pode identificar alteração dos reflexos, da coordenação, do equilíbrio, da marcha, dos nervos cranianos entre outras. A confirmação do diagnóstico é feita pela Ressonância Nuclear Magnética que mostra o defeito na junção entre o crânio e a região cervical.
Essa doença exige o acompanhamento com neurocirurgião, para que o tratamento cirúrgico seja indicado no momento oportuno, quando aparecem evidências da deterioração neurológica, progressão dos sintomas que se tornam incapacitantes e piora das alterações na Ressonância Magnética. A cirurgia é feita através de uma incisão na parte de trás da cabeça e do pescoço sob anestesia geral e visa a descompressão das estruturas nervosas e o restabelecimento da circulação do líquor. Essa cirurgia geralmente apresenta ótimos resultados.
Referências
- Izquierdo Martínez M and Avellaneda Fernández A. Enfermedades Raras: Un Enfoque Práctico. 2004, 511-12. http://iier.isciii.es/er/prg/er_bus2.asp?cod_enf=178
A síndrome de Arnold-Chiari, também chamada de má formação de Arnold-Chiari, refere-se a uma rara malformação de origem congênita do sistema nervoso central (SNC).
Esta condição é ocasionada por um deslocamento inferior das amígdalas cerebelares, passando pela abertura occipital situada na base do crânio, levando, em muitos casos, à hidrocefalia como consequência da obstrução da circulação do líquido cefalorraquidiano.
Foi descrita pela primeira vez pelo patologista austríaco Hans Chiari, no fim do século XIX. Este, por sua vez descreveu três tipos distintos desta desordem: tipo I, II e III. Posteriormente, outros estudiosos acrescentaram outro tipo à síndrome em questão, o tipo IV.
A síndrome do tipo I tipicamente é assintomática durante a infância. Quando há a presença de sintomas, caracteriza-se por cefaleia, dor de garganta, deambulação instável.
No tipo II pode ser encontrada uma mielomeningocele lombar ou uma herniação tonsilar abaixo do forame magno. Isso pode levar a paralisia abaixo do defeito espinhal.
A síndrome de Arnold-Chiari do tipo III está ligada a uma encefalocele occipital, contendo uma diversidade de tecidos neuroectodérmicos anormais. Neste tipo são observados os sintomas descritos no tipo I e II, além de déficit neurológico.
O tipo IV, posteriormente descrito, caracteriza-se pela ausência de desenvolvimento cerebelar, sendo este tipo incompatível com a vida.
Outras duas condições que comumente encontram-se relacionadas com esta desordem são as síndrome de Ehlers-Danlos e de Marfan.
O diagnóstico é feito com base no histórico e quadro clínico apresentado pelo paciente, sendo confirmado por exames de imagem, como ressonância magnética e/ou tomografia computadorizada. Investigação do fluxo do líquido cefalorraquidiano também pode ser útil para o diagnóstico.
O tratamento desta síndrome é feito com base nas manifestações. Quando há compressão da medula espinhal, é necessário realizar cirurgia para descomprimi-la. Em outros casos, a terapia física ou ocupacional pode ajudar a melhorar a coordenação motora, bem como a fonoaudiologia pode ser útil para melhorar a dicção do paciente.
Fontes:
http://en.wikipedia.org/wiki/Arnold%E2%80%93Chiari_malformation
http://jobaccess.gov.au/Advice/Disability/Pages/Arnold_Chiari_Syndrome.aspx
Esta condição é ocasionada por um deslocamento inferior das amígdalas cerebelares, passando pela abertura occipital situada na base do crânio, levando, em muitos casos, à hidrocefalia como consequência da obstrução da circulação do líquido cefalorraquidiano.
Foi descrita pela primeira vez pelo patologista austríaco Hans Chiari, no fim do século XIX. Este, por sua vez descreveu três tipos distintos desta desordem: tipo I, II e III. Posteriormente, outros estudiosos acrescentaram outro tipo à síndrome em questão, o tipo IV.
A síndrome do tipo I tipicamente é assintomática durante a infância. Quando há a presença de sintomas, caracteriza-se por cefaleia, dor de garganta, deambulação instável.
No tipo II pode ser encontrada uma mielomeningocele lombar ou uma herniação tonsilar abaixo do forame magno. Isso pode levar a paralisia abaixo do defeito espinhal.
A síndrome de Arnold-Chiari do tipo III está ligada a uma encefalocele occipital, contendo uma diversidade de tecidos neuroectodérmicos anormais. Neste tipo são observados os sintomas descritos no tipo I e II, além de déficit neurológico.
O tipo IV, posteriormente descrito, caracteriza-se pela ausência de desenvolvimento cerebelar, sendo este tipo incompatível com a vida.
Outras duas condições que comumente encontram-se relacionadas com esta desordem são as síndrome de Ehlers-Danlos e de Marfan.
O diagnóstico é feito com base no histórico e quadro clínico apresentado pelo paciente, sendo confirmado por exames de imagem, como ressonância magnética e/ou tomografia computadorizada. Investigação do fluxo do líquido cefalorraquidiano também pode ser útil para o diagnóstico.
O tratamento desta síndrome é feito com base nas manifestações. Quando há compressão da medula espinhal, é necessário realizar cirurgia para descomprimi-la. Em outros casos, a terapia física ou ocupacional pode ajudar a melhorar a coordenação motora, bem como a fonoaudiologia pode ser útil para melhorar a dicção do paciente.
Fontes:
http://en.wikipedia.org/wiki/Arnold%E2%80%93Chiari_malformation
http://jobaccess.gov.au/Advice/Disability/Pages/Arnold_Chiari_Syndrome.aspx

Malformação de Arnold-Chiari tipo 1 (Chiari 1)

Os sintomas costumam aparecer na fase adulta entre a terceira e quarta décadas de vida e os mais comuns são: dor cervical, dor de cabeça intensa, fraqueza muscular, dormência ou alteração da sensibilidade nos membros e dificuldade de equilíbrio. Outros sintomas que podem surgir são: vertigem, distúrbios visuais, zumbidos, dificuldade para engolir, palpitação, apnéia do sono, diminuição das habilidades motoras finas e fadiga crônica. O exame neurológico realizado pelo especialista auxilia na determinação do diagnóstico, uma vez que pode identificar alteração dos reflexos, da coordenação, do equilíbrio, da marcha, dos nervos cranianos entre outras. A confirmação do diagnóstico é feita pela Ressonância Nuclear Magnética que mostra o defeito na junção entre o crânio e a região cervical.
Essa doença exige o acompanhamento com neurocirurgião, para que o tratamento cirúrgico seja indicado no momento oportuno, quando aparecem evidências da deterioração neurológica, progressão dos sintomas que se tornam incapacitantes e piora das alterações na Ressonância Magnética. A cirurgia é feita através de uma incisão na parte de trás da cabeça e do pescoço sob anestesia geral e visa a descompressão das estruturas nervosas e o restabelecimento da circulação do líquor. Essa cirurgia geralmente apresenta ótimos resultados.
==================================================================
Síndrome de Down
Síndrome de Down ou Trissomia 21
Síndrome de Down ou Trissomia 21
História da síndrome de Down
Gisele Santos de Oliveira ¹
Meire Gomes ²
Até o momento, não há descrição de registro relacionável à SD antes do século XIX. A existência de uma síndrome com faciescaracterístico
foi oficialmente apresentada à comunidade científica pela primeira vez
por Edouard Onesimus Seguin, em 1846, mas não havia um estudo publicado
caracterizando uma pesquisa.
O
primeiro relato sobre a síndrome foi feito entre 1864 e 1866, pelo
médico inglês John Langdon Haydon Down, que trabalhava em uma clínica
para crianças com atraso neuropsicomotor, em Surrey, na Inglaterra. Ele
listou com clareza as características físicas similares que observou em
alguns filhos de mães acima de 35 anos de idade, descrevendo as crianças
como “amáveis e amistosas”. Influenciado pela Teoria da Evolução de
Charles Darwin, o médico explicou a síndrome estabelecendo uma teoria
étnica, sugerindo ser a síndrome um “estado regressivo da evolução”.
Após
a descrição de Down, os estudos sobre a causa da síndrome atribuiram-na
à tuberculose, à sífilis e ao hipotireoidismo, sendo os pacientes
considerados como “crianças inacabadas”.
Durante
o período que antecedeu a identificação da alteração cromossômica, os
pacientes foram rejeitados e mantidos sob regime hospitalar, em
condições precárias. O fim desse primeiro período da história da SD é
marcado por uma intolerância de raízes religiosas e culturais e coincide
com o Holocausto Judeu, um dos ícones do preconceito humano.
Em
1959, quase cem anos após a descrição do Dr. Down, os cientistas Jerome
LeJeune e Patricia Jacobs, trabalhando de forma independente,
determinaram a causa do até então “mongolismo”, como sendo a trissomia
do cromossomo 21.
A
trissomia do 21 foi a PRIMEIRA alteração cromossômica detectada na
espécie humana e dentro dos primeiros anos da década seguinte, seria
renomeada para Síndrome de Down. A descoberta da alteração cromossômica
marca o segundo período da história da SD, trazendo consigo uma fase
repaginada de interesse científico.
Em
1960, Polani descreveu casos de Translocação, isto é, partes do ou o
cromossomo 21 inteiro colado em outro cromossomo qualquer, e em 1961,
descreveu o primeiro caso de Mosaicismo, sendo duas linhagens celulares
possuindo diferentes padrões cromossômicos. No caso da SD, uma linhagem
celular com 46 (normal) cromossomos e outra com 47 (com a trissomia 21).
Nos
Estados Unidos, após uma revisão de termos cientifícos realizada em
1970, a denominação “mongolismo” foi abolida e a alteração foi
definitivamente denominada Síndrome de Down (Down Syndrome), em
homenagem ao médico que a descreveu pela primeira vez.
O
terceiro período da história da SD coincide com a onda de
reconhecimento dos direitos da criança e do adolescente, que foi tomando
conta de grande parte do mundo a partir das últimas décadas do século
XX, onde toda criança, independente de sexo, raça, cor, religião ou
capacidade mental teria direito a cuidados médicos e educação. Começou
assim, a fase do interesse científico aliado ao interesse educacional e
hoje, a institucionalização, a marginalização e a ignorância, enfim, vão
cedendo paulatinamente lugar ao seguimento inter-disciplinar humanizado
e especializado e a programas educacionais cada vez mais ricos,
pautados no conhecimento de que as pessoas com SD têm inúmeras
potencialidades e revelam-nas quando bem integradas à família e à
comunidade.
Hoje,
nossas crianças podem ter um desenvolvimento motor muito próximo às
crianças que não tem SD, falam, correm, brincam. Muitas aprendem a ler,
escrever; outras, a tocar piano; muitas praticam esportes, e tantas
outras dedicam-se a uma profissão ou às artes, conforme suas
potencialidades – que devem ser aproveitadas ao máximo.
O
objetivo da equipe, além de manter a saúde da criança, é impulsionar o
seu desenvolvimento e socialização. Nós, profissionais, pais, mães e
todos que estão à nossa volta podem ser alistados nesse exército do bem.
Basta querer. E sem desânimo, sempre.
AUTORAS:
¹
Médica pela Universidade Federal do Paraná, residência em pediatria
pela UFPR, residência em genética médica pela FCM/Unicamp, título de
especialista Genética Clínica pela Associação Médica Brasileira e
Sociedade Brasileira de Genética Clínica, mestrado Ciências Médicas –
concentração em Genética Médica pela FCM/Unicamp, doutoranda – Ciências
Médicas – concentração em Genética Médica pela FCM/Unicamp.
²
Médica pela Universidade Federal do Rio Grande do Norte, residência em
pediatria pela UFRN, título de especialista em Pediatria pela Associação
Médica Brasileira e Sociedade Brasileira de Pediatria; ex-médica da
rede estadual de assistência à pessoa com deficiência do Rio Grande do
Norte – Centro de Reabilitação Infantil; Curso de Formação em Perícia
Médica, pós-graduanda em Direito Previdenciário.
REFERÊNCIAS:
1. Down, JL. Observation on an ethnic classification of idiots. London Hospital. Clinical Lectures and Reports 1866;3:259-262
2.
Lejeune J, Gautier M, Turpin R. Etudes des chromosomes somatiques de
neuf enfants mongoliens. Comptes Rendues Hebdomadaires des Seances de
L’Academie des Sciences. Paris. 1959;248:602-603
3. Margotta, R. História Ilustrada da Medicina. Londres. Ed. Manole 1998; 8:152.
4. Mustacchi, Zan. Síndrome de Down. Brasil. CID Editora Ltda 1990; 1: 21
5.
Bartshaw, ML. A criança com deficiências do desenvolvimento. Clínicas
Pediátricas da América do Norte. Interlivros 1993; 567-568.
6. http://www.ufrgs.br/HCPA/gppg/crogen.htm
7. http://www.novanet.com.br/riodown/indown01.htm
8. http://www.medicina.ufmg.br/down/introducao.htm
9. http://geocities.yahoo.com.br/bazeggiobr/texto_sdown.htm
10. http://www.cromossomo21.hpg.ig.com.br/index.htm
11. http://www.terravista.pt/meco/4625/S%C3%ADndrome%20de%20Down.htm
12.
Online Mendelian Inheritance in Man, OMIM (TM). – McKusick-Nathans
Institute for Genetic Medicine, Johns Hopkins University (Baltimore, MD)
and National Center for Biotechnology Information, National Library of
Medicine (Bethesda, MD), 2006. World Wide Web URL:
http://www.ncbi.nlm.nih.gov/omim/.
13. Epstein, C. J. – Down syndrome, trisomy 21. In: Scriver, C. R.; Beaudet, A. L.; Sly, W. S.; Valle, D. : Metabolic Basis of Inherited Disease. New York: McGraw-Hill (pub.) 1989. 291-326p.
14. JONES KL – Smith’s recognizable patterns of human malformation. 5.ed. Phyladelphia. Saunders. 1997. 8-13p.
15. Mueller RF, Young ID. Emery’s Elements of Medical Genetics, Churchill Livingstone, New York, 9th ed, 1993.
16. Nora JJ et al. Medical Genetics: Principles and Practice, Lea & Febiger, 4th ed, 1993.
Diagnostico pré-natal da síndrome de Down
Autora: Gisele Santos Oliveira *
Atualmente, alguns métodos são empregados para a triagem de SD durante a gestação:
- Medida da translucência nucal (TN)
- Teste triplo ou tri-teste
- Teste de risco fetal
- Biópsia de vilo corial
- Amniocentese
Medida da translucência nucal (TN)
Deve
ser lembrada a importância do acompanhamento gestacional, por meio do
ultra-som obstétrico, com o que são chamados “marcadores biofísicos”,
como a medida do úmero e sua relação com o diâmetro biparietal, o
comprimento céfalo-caudal e a translucência nucal.
A TN, medida entre as 10a e 12a
semanas, foi associada, em diversos estudos, a um risco elevado para
SD. Essa medida é a espessura máxima da translucência subcutânea, espaço
compreendido entre a pele e os tecidos moles, presente na região da
nuca. Ela é produzida pelo acúmulo de líquidos nesse local e, ao
ultra-som, é demonstrada como uma imagem anecóica (escura).
Teste triplo ou tri-teste
O
teste triplo, ou tri-teste, refere-se à dosagem de três marcadores
bioquímicos do soro da gestante: a alfa-fetoproteína (αFP), o estriol
não conjugado (uE3) e a gonadotrofina coriônica humana livre (β-HCG).
A
αFP é uma proteína produzida pelo fígado do feto e começa a estar
presente na circulação materna a partir da 14ª semana gestacional. O uE3
é um estrogênio cuja síntese é determinada pela associação entre o
fígado e a supra-renal fetais com a placenta. O β-HCG é produzido pela
placenta e detectado na circulação materna a partir do 7º dia
pós-concepção, aumentando progressivamente até a 10ª semana gestacional e
regredindo lentamente até o final da gestação.
A
dosagem dessas três substâncias identifica, além da SD e trissomia 18
(síndrome de Edwards), fetos com risco de defeitos de fechamento do tubo
neural (DFTN), como espinha bífida com mielomeningocele, encefalocele,
entre outras.
As gestantes de crianças portadoras de SD possuem níveis muito baixos de αFP e uE3 e taxas elevadas de β-HCG, quando comparadas com gestantes de fetos sem anomalias.
Dessa
forma, o período indicado para a coleta do sangue materno é o segundo
trimestre, mais precisamente, o período compreendido entre as semanas
gestacionais de 12 a 22 semanas e 06 dias, podendo, assim, fornecer uma
indicação de alteração fetal.
Atualmente,
tem sido dada preferência ao teste integrado ou avaliação do risco
fetal, onde, já no primeiro trimestre é realizada a medida da TN,
juntamente com a dosagem da proteína plasmática A associada à gravidez
(PAPP-A), que funciona como um regulador na formação e crescimento de
diversos sistemas e aparelhos humanos, como o sistema cardiovascular e o
aparelho reprodutor e, no segundo trimestre, a avaliação é
complementada com o tri-teste.
Biópsia de vilo corial e amniocentese
Tanto
a biópsia de vilo corial quanto a amniocentese tem indicações precisas e
são métodos mais reservados, por serem invasivos ao ambiente fetal. São
de indicação formal quando a gestante tem mais de 34 anos, quando
existe uma criança prévia com SD ou qualquer outra cromossomopatia e
quando um dos pais é portador de uma translocação equilibrada. Ainda,
quando após a realização da avaliação do risco fetal, houver resultado
indicativo de anomalias fetais.
Assim,
diante de um valor alterado, indica-se o cariótipo fetal, através de
cultura das vilosidades coriônicas ou de células em suspensão no líquido
amniótico, coletadas pela biópsia de vilo corial ou pela amniocentese.
Esses tecidos são provenientes de folhetos embrionários produzidos pela
divisão do zigoto, tendo, assim, o mesmo material genético do feto, e
podendo ser coletados e examinados, fornecendo um resultado fidedigno.
A
biópsia de vilo corial, realizada a partir da 7ª semana gestacional,
pode ser tanto transcervical quanto transabdominal e consiste,
basicamente, na inserção intra-uterina de um catéter que tenha em seu
interior um mandril que possa lhe dar a direção. Por ser um período em
que o córion começa a se diferenciar (córion frondoso) para a produção
da placenta, com um alto índice mitótico (divisão celular) é, portanto, a
área da qual será coletado o material.
A
amniocentese, realizada a partir da décima quarta semana, é um dos
métodos mais difundidos para a obtenção de material fetal com finalidade
de diagnóstico pré-natal de alterações genéticas. A segurança e o baixo
índice de complicações decorrentes da técnica fizeram com que ela se
tornasse rotina na maioria dos serviços.
Considerando
as duas técnicas, vale a pena ser lembrado que a monitorização fetal,
por meio de ultra-som é de indicação formal, antes, durante e após o
procedimento.
Recentemente,
técnicas de imunofluorescência têm feito a detecção de células fetais
circulantes no sangue materno para análise de cariótipo e molecular para
o diagnóstico do feto.
* Médica
pela Universidade Federal do Paraná, residência em pediatria pela UFPR,
residência em genética médica pela FCM/Unicamp, título de especialista
Genética Clínica pela Associação Médica Brasileira e Sociedade
Brasileira de Genética Clínica, mestrado Ciências Médicas – concentração
em Genética Médica pela FCM/Unicamp, doutoranda – Ciências Médicas –
concentração em Genética Médica pela FCM/Unicamp.
Referências
JONES KL – Smith’s recognizable patterns of human malformation. 5.ed. Phyladelphia. Saunders. 1997. 8-13p.
Online
Mendelian Inheritance in Man, OMIM (TM). – McKusick-Nathans Institute
for Genetic Medicine, Johns Hopkins University (Baltimore, MD) and
National Center for Biotechnology Information, National Library of
Medicine (Bethesda, MD), 2006. World Wide Web URL:
http://www.ncbi.nlm.nih.gov/omim/.
Harper PS. Practical Genetic Counseling, Butterworth-Heinemann, Oxford, 4th ed, 1994.
Mueller RF, Young ID. Emery’s Elements of Medical Genetics, Churchill Livingstone, New York, 9th ed, 1993.
Nora JJ et al. Medical Genetics: Principles and Practice, Lea & Febiger, 4th ed, 1993.
ACACIO GL, BARINI R, PINTO JUNIOR W et al. Nuchal translucency: na ultrasound marker for fetal chromosomal abnormalities. Sao Paulo Med J/Rev Paul Med 2001; 119(1):19-23
Pinto Jr W et al. 1987. Diagnóstico pré-natal e genético.Neurologia Infantil. Belo Horizonte, 74-82 pp
Pinto Jr W. Diagnóstico pré-natal. Ciência & Saúde Coletiva, 7(1). Rio de Janeiro, 139-157, 2002.
Grillo
LB, Acacio GL, Barini R, Pinto W Jr, Bertuzzo CS. Mutations in the
methylene-tetrahydrofolate reductase gene and Down syndrome.
Cad Saude Publica. 2002 Nov-Dec;18(6):1795-7.
Cad Saude Publica. 2002 Nov-Dec;18(6):1795-7.
Aconselhamento Genético na síndrome de Down
Autora: Maria Ione Costa *
O
diagnóstico da Síndrome de Down pode, quase sempre com segurança, ser
realizado apenas com os sinais clínicos; entretanto para o
aconselhamento genético (AG), deve-se estudar o cariótipo, pois se o
caso for de trissomia por translocação, pode ser que essa tenha sido
herdada de um dos progenitores, portador da translocação em estado
equilibrado. Se a trissomia for simples (sem translocação) ou se houver
mosaicismo 46/47, o risco de recorrência é fixado em: 1% para mulheres
abaixo de 35 anos que já tiveram um afetado com SD, de 2% até os 40
anos, 3% até os 43 e 5% daí em diante.
Na
prática, o risco que corre qualquer casal tomado ao acaso de que uma
criança nasça com defeito ou doença genética é avaliado em torno de 3%;
esse risco geral é acrescido ao específico para cada idade na SD,
oferecendo-se um risco final.
Em
casos de translocação esporádicas (presentes somente nos pacientes com
SD), o AG é semelhante ao do causado por trissomia simples do cromossomo
21; entretanto quando a mãe é portadora da translocação D/21 ou 21/22, o
risco empírico de recorrência é estimado em 10 a 20% e quando o pai é
portador, o risco empírico cai para 5 a 10%. Se a translocação
equilibrada presente me um dos genitores for 21/21, o risco de
recorrência é de 100%. Por isso são importantes as técnicas de
identificação dos cromossomos por bandeamento, pois só assim se pode
diferenciar entre os tipos de diagnósticos citogenéticos na SD.
in http://espacodown.wordpress.com/
A
síndrome de Down (SD) é uma alteração genética produzida pela presença
de um cromossomo a mais, o par 21, por isso também conhecida como trissomia 21.
A SD
foi descrita em 1866 por John Langdon Down. Esta alteração genética
afeta o desenvolvimento do individuo, determinando algumas
características físicas e cognitivas. A maioria das pessoas com SD
apresenta a denominada trissomia 21 simples, isto significa que
um cromossomo extra está presente em todas as células do organismo,
devido a um erro na separação dos cromossomos 21 em uma das células dos
pais. Este fenômeno é conhecido como disfunção cromossômica. Existem
outras formas de SD como, por exemplo: mosaico, quando a trissomia está
presente somente em algumas células, e por translocação, quando o
cromossomo 21 está unido a outro cromossomo.
O
diagnóstico da SD se realiza mediante o estudo cromossômico
(cariótipo), através do qual se detecta a presença de um cromossomo 21 a
mais. Este tipo de análise foi utilizado pela primeira vez em 1958 por
Jerome Lejeune.
Não
se conhece com precisão os mecanismos da disfunção que causa a SD, mas
está demonstrado cientificamente que acontece igualmente em qualquer
raça, sem nenhuma relação com o nível cultural, social, ambiental,
econômico, etc. Há uma maior
probabilidade da presença de SD em relação à idade materna, e isto é
mais freqüente a partir dos 35 anos, quando os riscos de se gestar um
bebê com SD aumenta de forma progressiva. Paradoxalmente, o nascimento
de crianças com SD é mais freqüente entre mulheres com menos de 35 anos,
isto se deve ao fato de que mulheres mais jovens geram mais filhos e
também pela influência do diagnóstico pré natal,que é oferecido
sistematicamente às mulheres com mais de 35 anos.
Como a SD é uma alteração cromossômica, é possível realizar um diagnóstico pré natal utilizando diversos exames clínicos como, por exemplo, a amniocentese (pulsão transabdominal do
liquido amniótico entre as semanas 14 e 18 de gestação) ou a biópsia do
vilo corial (coleta de um fragmento da placenta). Ambos os exames
diagnosticam a SD e outras cromossopatias.
Recentemente
a prática médica tem incorporado métodos para a determinação do risco
de ter um filho com SD, como por exemplo, o exame bioquímico, que se
realiza mediante a avaliação dos níveis de substâncias químicas no
sangue materno alteradas no caso da SD. Este exame se realiza entre a
semana 14 e 17. A ultrassonografia também pode colaborar para detectar a
SD, através dos marcadores ecográficos, principalmente da prega nucal,
que pode ser medida a partir da décima semana de gestação. Estas últimas
intervenções não são consideradas diagnósticas, para isso é necessário
realizar os exames mencionados em primeiro lugar.
Embora
as alterações cromossômicas da SD sejam comuns a todas as pessoas, nem
todas apresentam as mesmas características, nem os mesmos traços
físicos, tampouco as malformações. A única característica comum a todas
as pessoas é o déficit intelectual. Não existem graus de SD; a variação
das características e personalidades entre uma pessoa e outra é a mesma
que existe entre as pessoas que não tem SD.
Cerca
de 50% das crianças com SD apresentam problemas cardíacos, algumas
vezes graves, necessitando de cirurgia nos primeiros anos de vida.
A
intervenção médica pode acontecer com a finalidade principal de
prevenção dos problemas de saúde que podem aparecer com maior freqüência
na SD. Queremos destacar que a SD não é uma doença e sim uma alteração
genética, que pode gerar problemas médicos associados.
Devemos olhar a pessoas com SD em sua singularidade, para que possa ter um pleno desenvolvimento enquanto sujeito.
n http://www.fsdown.org.br/
***********************************************************
Síndrome de Warkany ou Trissomia 8
Síndrome de Warkany ou Trissomia 8



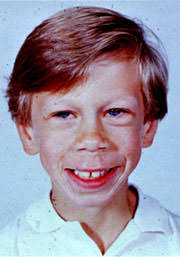
O QUE É? A SÍNDROME DE WILLIAMS TAMBÉM CONHECIDA COMO SÍNDROME WILLIAMS-BEUREN É UMA DESORDEM GENÉTICA QUE, TALVEZ, POR SER RARA, FREQÜENTEMENTE NÃO É DIAGNOSTICADA. SUA TRANSMISSÃO NÃO É GENÉTICA. O NOME DESTA SÍNDROME VEM DO MÉDICO, DR. J.C.P. WILLIAMS QUE A DESCREVEU EM 1961 NA NOVA ZELÂNDIA E PELO DR. A. J. BEUREN DA ALEMANHA EM 1962.
ACOMETENDO AMBOS OS SEXOS, NA MAIORIA DOS CASOS INFANTIS (PRIMEIRO ANO DE VIDA), AS CRIANÇAS TÊM DIFICULDADE DE SE ALIMENTAR, FICAM IRRITADAS FACILMENTE E CHORAM MUITO.
A SÍNDROME DE WILLIAMS É UMA DOENÇA CARACTERIZADA POR "FACE DE GNOMO OU FADINHA”, NARIZ PEQUENO E EMPINADO, CABELOS ENCARACOLADOS, LÁBIOS CHEIOS, DENTES PEQUENOS E SORRISO FREQÜENTE. ESTAS CRIANÇAS NORMALMENTE TÊM PROBLEMAS DE COORDENAÇÃO E EQUILÍBRIO, APRESENTANDO UM ATRASO PSICOMOTOR. SEU COMPORTAMENTO É SOCIÁVEL E COMUNICATIVO EMBORA UTILIZEM EXPRESSÕES FACIAIS, CONTATOS VISUAIS E GESTOS EM SUA COMUNICAÇÃO.
EMBORA COMECEM A FALAR TARDE, POR VOLTA DOS 18 MESES, DEMONSTRAM FACILIDADE PARA APRENDER RIMAS E CANÇÕES, DEMONSTRANDO MUITA SENSIBILIDADE MUSICAL E CONCOMITANTEMENTE BOA MEMÓRIA AUDITIVA.
SEU DESENVOLVIMENTO MOTOR É MAIS LENTO. DEMORAM A ANDAR, E TEM GRANDE DIFICULDADE EM EXECUTAR TAREFAS QUE NECESSITEM DE COORDENAÇÃO MOTORA TAIS COMO: CORTAR PAPEL, DESENHAR, ANDAR DE BICICLETA, AMARRAR O SAPATO ETC..
TRATAMENTO E PREVENÇÃO DAS COMPLICAÇÕES.
É MUITO IMPORTANTE IDENTIFICAR PORTADORES DESTA SÍNDROME LOGO NA PRIMEIRA INFÂNCIA, POIS, TEM INFLUÊNCIA EM DIVERSAS PARTES DO DESENVOLVIMENTO COGNITIVO, COMPORTAMENTAL E MOTOR.
AS MEDIDAS PREVENTIVAS DEVEM-SE INICIAR LOGO APÓS O DIAGNÓSTICO COM UM ESTUDO MINUCIOSO PARA DESCARTE DE ANOMALIAS DO CORAÇÃO E RINS. É NECESSÁRIO MONITORAR FREQÜENTEMENTE A HIPERTENSÃO ARTERIAL, INCLUINDO A AVALIAÇÃO DA TENSÃO ARTERIAL NOS QUATRO MEMBROS.
A OTITE CRÔNICA EXIGE AVALIAÇÕES AUDITIVAS FREQÜENTES E QUANDO NECESSÁRIO O ENVIO PARA UMA CONSULTA DE OTORRINOLARINGOLOGIA. O TRATAMENTO DE PROBLEMAS DENTÁRIOS NECESSITA DA PROFILAXIA DA ENDOCARDITE. FACE ÀS INFECÇÕES URINÁRIAS FREQÜENTES TORNA-SE NECESSÁRIO AVALIAR A FUNÇÃO RENAL PERIODICAMENTE E REALIZAR UM ESTUDO MINUCIOSO NA INFÂNCIA E NA ADOLESCÊNCIA.
NA ADOLESCÊNCIA, PARA ALÉM DE SE MANTER A VIGILÂNCIA DOS SISTEMAS JÁ DESCRITOS, DEVE-SE PESQUISAR A PRESENÇA DE ESCOLIOSE E CONTRATURA DAS ARTICULAÇÕES. OS PROBLEMAS ALIMENTARES OBSERVADOS NOS MAIS NOVOS SÃO ULTRAPASSADOS, SENDO A OBESIDADE ENCONTRADA EM 29% DOS ADULTOS. O COMPORTAMENTO E APROVEITAMENTO ESCOLAR, QUANDO PROBLEMÁTICOS CARECEM DE MEDIDAS DE APOIO. A ANSIEDADE PODE ESTAR ASSOCIADA À ÚLCERA PÉPTICA E A LITÍASE BILIAR É UM DIAGNÓSTICO POSSÍVEL EM DOENTES COM DORES ABDOMINAIS
PERSONALIDADE E COMPORTAMENTO
NAS CRIANÇAS PORTADORAS DESTA SÍNDROME É GRANDE A SOCIABILIDADE, ENTUSIASMO, GRANDE SENSIBILIDADE, TEM UMA MEMÓRIA FANTÁSTICA PARA PESSOAS, NOMES E LOCAL; ANSIEDADE MEDO DE ALTURAS, PREOCUPAÇÃO EXCESSIVA COM DETERMINADOS ASSUNTOS OU OBJETOS, DISTÚRBIOS DO SONO, CONTROLE DO ESFÍNCTER É NORMAL CRIANÇAS COM ESTA SÍNDROME SEREM AMIGAS DE ADULTOS E PROCURAREM A COMPANHIA DELES AO MESMO TEMPO TEM DIFICULDADE EM FAZER AMIZADES OUTRAS CRIANÇAS DA SUA IDADE. MUITAS CRIANÇAS COM ESTA SÍNDROME DEMONSTRAM MEDO AO ESCUTAREM RUÍDOS DE BATER PALMAS, LIQUIDIFICADOR, AVIÃO, ETC., POR SEREM HIPERSENSÍVEIS AO SOM.

Foi
descoberta pelo pediatra austríaco-americano, Joseph warkany,
caracterizando-se pela presença de um cromossoma 8 adicional. Esta
anomalia é geralmente mortal, resultando em abortos espontâneos.
Existem
três fases de desenvolvimento propícias à mutação: o período da
blástula, que corresponde às três primeiras semanas; o período
embrionário, que se localiza entre a 4ª e a 12ª semana, aquando da
formação dos tecidos e órgãos e o período fetal, desde a 13ª à 40ª
semana, durante o crescimento do feto.
Fenotipicamente
o síndroma manifesta-se com diferentes graus de dimorfismo, de leve a
intenso, caracterizando-se principalmente por: espasmos; anomalias
vertebrais; anomalias renais; défice cognitivo, moderado a grave; atraso
no desenvolvimento psicomotor; pélvis estreita; postura anormal e
grande afastamento dos dedos dos pés.
in http://astrissomias.blogspot.pt/2011/05/trissomia-8.html
A síndrome de Warkany ou trissomia 8
caracteriza-se pelo comprometimento do desenvolvimento das estruturas
mentais, conformação facial relativamente comum entre os portadores,
rótulas (articulação dos joelhos) ausentes ou displásicas, contracções
espasmódicas, sulcos nas plantas dos pés e nas palmas das mãos o que
provoca a postura distintiva anormal do dedo do pé, anomalia vertebral,
pélvis estreito, anomalias ureteral-renal, etc.
in http://iguais-na-diferenca.blogspot.pt/2008/02/sndrome-de-warkany-ou-trissomia-8.html
A
trissomia 8 em mosaico é uma anomalia cromossómica definida pela
presença de três cópias do cromossoma 8 em algumas células do organismo.
É caracterizada por dismorfismo facial, atraso mental ligeiro e
anomalias das articulações, urinárias cardíacas e esqueléticas.
A incidência anual varia entre 1/25,000 e 1/50,000. Os indivíduos do sexo masculino são mais frequentemente afetados que os do feminino (razão 5:1).
As características dismórficas facial são ligeiras e incluem alongamento do crânio (escafocefalia), fronte prominente, hipertelorismo, olhos encovados (50%), nariz largo e antevertido (60%), micrognatia (com eversão do lábio inferior), orelhas displásicas grandes com anti-hélices prominentes e lóbulos grandes. As características adicionais incluem: agenesia do corpo caloso, palato alto e arqueado ou fenda do palato (8%), pescoço curto e largo, alta estatura, tronco fino alongado, ombros e pélvis estreitos. São frequentes as anomalias urinárias (hidronefrose, refluxo ureteral) e cardíacas e dos grandes vasos (40% e 25% respetivamente). São também frequentemente observadas camptodactilia (70%), artrogripose das articulações (agravando com o tempo), pregas palmares (nos latentes) e plantares profundas (75%), rótula ausente ou hipoplásica, malformações vertebrais (65%: anomalias de segmentação, anomalias costais, escoliose... ) bem como opacidade da córnea e estrabismo. A maioria dos doentes apresenta atraso mental ligeiro (QI entre 50 e 75), com alguns doentes tendo inteligência normal.
A trissomia 8 em mosaico é o resultado de um evento pós-zigótico (erro na segregação do cromossoma durante a mitose num feto com um cariótipo normal ou correção espontânea da trissomia 8). A trissomia 8 completa é causada por um erro na segregação do cromossoma durante a meiose e resulta frequentemente em abortamento durante o primeiro trimestre. Quando, excecionalmente, o feto sobrevive, apresenta o mesmo fenótipo da trissomia em mosaico. Os doentes com isocromossoma 8p (tetrassomia do braço curto (p) do cromossoma 8) têm o mesmo fenótipo que os doentes com a trissomia 8p (ver este termo).
O diagnóstico é baseado na análise do cariótipo.
O diagnóstico pré-natal é possível através de estudos citogenéticos. A agenesia do corpo caloso é o critério ecográfico pré-natal mais importante para o diagnóstico do síndrome.
O aconselhamento genético é tranquilizador, já que a trissomia 8 é quase sempre um evento de novo e apresenta um baixo risco de recorrência.
O seguimento requer uma abordagem multidisciplinar. Em alguns casos, pode ser proposta cirurgia cardíaca. A trissomia 8 em mosaico parece predispor para tumor de Wilms, mielodisplasias e leucemia mielóide (ver estes termos). Alguns doentes com trissomia 8 em mosaico tiveram filhos.
Na ausência de malformações graves, a esperança média de vida é normal.
A incidência anual varia entre 1/25,000 e 1/50,000. Os indivíduos do sexo masculino são mais frequentemente afetados que os do feminino (razão 5:1).
As características dismórficas facial são ligeiras e incluem alongamento do crânio (escafocefalia), fronte prominente, hipertelorismo, olhos encovados (50%), nariz largo e antevertido (60%), micrognatia (com eversão do lábio inferior), orelhas displásicas grandes com anti-hélices prominentes e lóbulos grandes. As características adicionais incluem: agenesia do corpo caloso, palato alto e arqueado ou fenda do palato (8%), pescoço curto e largo, alta estatura, tronco fino alongado, ombros e pélvis estreitos. São frequentes as anomalias urinárias (hidronefrose, refluxo ureteral) e cardíacas e dos grandes vasos (40% e 25% respetivamente). São também frequentemente observadas camptodactilia (70%), artrogripose das articulações (agravando com o tempo), pregas palmares (nos latentes) e plantares profundas (75%), rótula ausente ou hipoplásica, malformações vertebrais (65%: anomalias de segmentação, anomalias costais, escoliose... ) bem como opacidade da córnea e estrabismo. A maioria dos doentes apresenta atraso mental ligeiro (QI entre 50 e 75), com alguns doentes tendo inteligência normal.
A trissomia 8 em mosaico é o resultado de um evento pós-zigótico (erro na segregação do cromossoma durante a mitose num feto com um cariótipo normal ou correção espontânea da trissomia 8). A trissomia 8 completa é causada por um erro na segregação do cromossoma durante a meiose e resulta frequentemente em abortamento durante o primeiro trimestre. Quando, excecionalmente, o feto sobrevive, apresenta o mesmo fenótipo da trissomia em mosaico. Os doentes com isocromossoma 8p (tetrassomia do braço curto (p) do cromossoma 8) têm o mesmo fenótipo que os doentes com a trissomia 8p (ver este termo).
O diagnóstico é baseado na análise do cariótipo.
O diagnóstico pré-natal é possível através de estudos citogenéticos. A agenesia do corpo caloso é o critério ecográfico pré-natal mais importante para o diagnóstico do síndrome.
O aconselhamento genético é tranquilizador, já que a trissomia 8 é quase sempre um evento de novo e apresenta um baixo risco de recorrência.
O seguimento requer uma abordagem multidisciplinar. Em alguns casos, pode ser proposta cirurgia cardíaca. A trissomia 8 em mosaico parece predispor para tumor de Wilms, mielodisplasias e leucemia mielóide (ver estes termos). Alguns doentes com trissomia 8 em mosaico tiveram filhos.
Na ausência de malformações graves, a esperança média de vida é normal.
Editor(es)
- Prof Alain VERLOES
in http://www.orpha.net/consor/cgi-bin/index.php
A
síndrome de Warkany ou trissomia 8 é uma anomalia cromossômica,
descoberta pelo pediatra austríaco-estadunidense Joseph Warkany.
Caracteristicas
A
síndrome de Warkany se caracteriza pelo comprometimento do
desenvolvimento das estruturas mentais, conformação facial relativamente
comum entre os portadores, rótulas ausentes ou displásicas, contrações
espasmódicas, sulcos nas plantas dos pés e nas palmas das mãos o que
provoca a postura distintiva anormal do dedo do pé, anomalia vertebral,
pélvis estreito, anomalias ureteral-renal, ou outras anomalias.
in http://biogeneticarau.blogspot.pt/2009/02/sindrome-de-warkany.html
SÍNDROME DE WILLIAMS
O QUE É? A SÍNDROME DE WILLIAMS TAMBÉM CONHECIDA COMO SÍNDROME WILLIAMS-BEUREN É UMA DESORDEM GENÉTICA QUE, TALVEZ, POR SER RARA, FREQÜENTEMENTE NÃO É DIAGNOSTICADA. SUA TRANSMISSÃO NÃO É GENÉTICA. O NOME DESTA SÍNDROME VEM DO MÉDICO, DR. J.C.P. WILLIAMS QUE A DESCREVEU EM 1961 NA NOVA ZELÂNDIA E PELO DR. A. J. BEUREN DA ALEMANHA EM 1962.
ACOMETENDO AMBOS OS SEXOS, NA MAIORIA DOS CASOS INFANTIS (PRIMEIRO ANO DE VIDA), AS CRIANÇAS TÊM DIFICULDADE DE SE ALIMENTAR, FICAM IRRITADAS FACILMENTE E CHORAM MUITO.
A SÍNDROME DE WILLIAMS É UMA DOENÇA CARACTERIZADA POR "FACE DE GNOMO OU FADINHA”, NARIZ PEQUENO E EMPINADO, CABELOS ENCARACOLADOS, LÁBIOS CHEIOS, DENTES PEQUENOS E SORRISO FREQÜENTE. ESTAS CRIANÇAS NORMALMENTE TÊM PROBLEMAS DE COORDENAÇÃO E EQUILÍBRIO, APRESENTANDO UM ATRASO PSICOMOTOR. SEU COMPORTAMENTO É SOCIÁVEL E COMUNICATIVO EMBORA UTILIZEM EXPRESSÕES FACIAIS, CONTATOS VISUAIS E GESTOS EM SUA COMUNICAÇÃO.
EMBORA COMECEM A FALAR TARDE, POR VOLTA DOS 18 MESES, DEMONSTRAM FACILIDADE PARA APRENDER RIMAS E CANÇÕES, DEMONSTRANDO MUITA SENSIBILIDADE MUSICAL E CONCOMITANTEMENTE BOA MEMÓRIA AUDITIVA.
SEU DESENVOLVIMENTO MOTOR É MAIS LENTO. DEMORAM A ANDAR, E TEM GRANDE DIFICULDADE EM EXECUTAR TAREFAS QUE NECESSITEM DE COORDENAÇÃO MOTORA TAIS COMO: CORTAR PAPEL, DESENHAR, ANDAR DE BICICLETA, AMARRAR O SAPATO ETC..
TRATAMENTO E PREVENÇÃO DAS COMPLICAÇÕES.
É MUITO IMPORTANTE IDENTIFICAR PORTADORES DESTA SÍNDROME LOGO NA PRIMEIRA INFÂNCIA, POIS, TEM INFLUÊNCIA EM DIVERSAS PARTES DO DESENVOLVIMENTO COGNITIVO, COMPORTAMENTAL E MOTOR.
AS MEDIDAS PREVENTIVAS DEVEM-SE INICIAR LOGO APÓS O DIAGNÓSTICO COM UM ESTUDO MINUCIOSO PARA DESCARTE DE ANOMALIAS DO CORAÇÃO E RINS. É NECESSÁRIO MONITORAR FREQÜENTEMENTE A HIPERTENSÃO ARTERIAL, INCLUINDO A AVALIAÇÃO DA TENSÃO ARTERIAL NOS QUATRO MEMBROS.
A OTITE CRÔNICA EXIGE AVALIAÇÕES AUDITIVAS FREQÜENTES E QUANDO NECESSÁRIO O ENVIO PARA UMA CONSULTA DE OTORRINOLARINGOLOGIA. O TRATAMENTO DE PROBLEMAS DENTÁRIOS NECESSITA DA PROFILAXIA DA ENDOCARDITE. FACE ÀS INFECÇÕES URINÁRIAS FREQÜENTES TORNA-SE NECESSÁRIO AVALIAR A FUNÇÃO RENAL PERIODICAMENTE E REALIZAR UM ESTUDO MINUCIOSO NA INFÂNCIA E NA ADOLESCÊNCIA.
NA ADOLESCÊNCIA, PARA ALÉM DE SE MANTER A VIGILÂNCIA DOS SISTEMAS JÁ DESCRITOS, DEVE-SE PESQUISAR A PRESENÇA DE ESCOLIOSE E CONTRATURA DAS ARTICULAÇÕES. OS PROBLEMAS ALIMENTARES OBSERVADOS NOS MAIS NOVOS SÃO ULTRAPASSADOS, SENDO A OBESIDADE ENCONTRADA EM 29% DOS ADULTOS. O COMPORTAMENTO E APROVEITAMENTO ESCOLAR, QUANDO PROBLEMÁTICOS CARECEM DE MEDIDAS DE APOIO. A ANSIEDADE PODE ESTAR ASSOCIADA À ÚLCERA PÉPTICA E A LITÍASE BILIAR É UM DIAGNÓSTICO POSSÍVEL EM DOENTES COM DORES ABDOMINAIS
PERSONALIDADE E COMPORTAMENTO
NAS CRIANÇAS PORTADORAS DESTA SÍNDROME É GRANDE A SOCIABILIDADE, ENTUSIASMO, GRANDE SENSIBILIDADE, TEM UMA MEMÓRIA FANTÁSTICA PARA PESSOAS, NOMES E LOCAL; ANSIEDADE MEDO DE ALTURAS, PREOCUPAÇÃO EXCESSIVA COM DETERMINADOS ASSUNTOS OU OBJETOS, DISTÚRBIOS DO SONO, CONTROLE DO ESFÍNCTER É NORMAL CRIANÇAS COM ESTA SÍNDROME SEREM AMIGAS DE ADULTOS E PROCURAREM A COMPANHIA DELES AO MESMO TEMPO TEM DIFICULDADE EM FAZER AMIZADES OUTRAS CRIANÇAS DA SUA IDADE. MUITAS CRIANÇAS COM ESTA SÍNDROME DEMONSTRAM MEDO AO ESCUTAREM RUÍDOS DE BATER PALMAS, LIQUIDIFICADOR, AVIÃO, ETC., POR SEREM HIPERSENSÍVEIS AO SOM.
in http://webleones.home.sapo.pt/williams.html
Síndrome de Williams
Informação adaptada de Preventive management of children with congenital anomalies and syndromes, Wilson GN, Cooley WC
Williams,
Beuren e colaboradores descreveram em 1961 e 1962, respectivamente, um
síndrome caracterizado por estenose supravalvular da aorta
(estreitamento da aorta acima da sua válvula) e fácies invulgar. A
estenose nem sempre está presente e pode haver outras alterações
associadas, nomeadamente a hipercalcémia neonatal (que em conjunto com
cardiopatia sugere esta situação) e o atraso mental.
Incidência, Etiologia e Diagnóstico
A
incidência desta patologia varia entre 1 em 20000 a 1 em 50000
nados-vivos. Em cerca de 90% dos doentes estão presentes delecções na
região 7q11 do cromossoma 7 que contêm o gene da elastina. Nos restantes
doentes o diagnóstico é clínico. A ausência de uma cópia do gene da
elastina é responsável pela estenose supravalvular aórtica e outras
alterações do tecido conjuntivo. Outras manifestações poderão ser
explicadas pela delecção de genes adjacentes a este. Existem outras
patologias genéticas que condicionam anomalias semelhantes e que terão
que ser excluídas.
O diagnóstico neonatal é habitualmente difícil, excepto em situações em que se detectam níveis de cálcio alto, uma vez que manifestações tal como o fácies característico, o aspecto da íris, o estrabismo, os lábios grossos e o sulco naso-labial longo se tornam habitualmente evidentes numa fase mais tardia.
O diagnóstico neonatal é habitualmente difícil, excepto em situações em que se detectam níveis de cálcio alto, uma vez que manifestações tal como o fácies característico, o aspecto da íris, o estrabismo, os lábios grossos e o sulco naso-labial longo se tornam habitualmente evidentes numa fase mais tardia.
Evolução
Os
primeiros meses cursam com cólicas abdominais e dificuldades
alimentares. Anomalias renais e espasmos do choro também podem ocorrer
nesta fase. Mais tarde, alterações visuais subtis e dificuldades na
aprendizagem tornam-se mais pertinentes.
As características faciais alteram-se consideravelmente com a idade. A esperança média de vida pode, devido às anomalias cardiovasculares e renais, estar ligeiramente diminuído, mas muitos estudos têm demonstrado a presença de excelente qualidade de vida em adultos com o síndrome de Williams. Apesar da estenose supravalvular aórtica e pulmonar, assim como a estenose da artéria renal e consequente hipertensão poderem causar problemas, nas crianças são mais os problemas comportamentais do que os do foro médico que preocupam os pais. Por um lado são indivíduos com personalidades sociais e de aspecto feliz, no entanto, a sua hiperactividade, labilidade emocional e ansiedade excessiva são particularidades que dificultam o seu relacionamento. A hiperacusia está frequentemente presente, podendo provocar reacções extremas a sons tais como o toque de campainhas e o ruído do cortador de relva, o que complica a terapêutica das alterações do comportamento. A microcefalia (cabeça pequena) e atraso do desenvolvimento são habituais e 59% destas crianças têm um QI inferior a 70. No entanto, muitas crianças cumprem os parâmetros apropriados à idade em diversas áreas tais como o reconhecimento visual, a linguagem expressiva e repetição de palavras.
Os mecanismos responsáveis pela hipercalcémia não estão completamente elucidados, podendo estar relacionados com uma resposta fisiológica diminuída a níveis normais de cálcio na alimentação e poderá contribuir para coarctação da aorta adquirida. Complicações mais tardias incluem o prolapso da válvula mitral, a úlcera péptica, a litíase biliar, a obesidade, as infecções urinárias e a diabetes mellitus. A hipercalcémia pode continuar a manifestar-se na vida adulta, por vezes resultando na excreção paradoxal da paratormona. Na 2ª e 3ª décadas a hipertensão arterial provocada pela calcificação dos rins e estenose das artérias renais torna-se mais frequente. Devido a estenoses das artérias cerebrais, alguns doentes sofrem acidentes vasculares cerebrais quando ainda jovens. Malformações do tracto urinário, incluindo divertículos vesicais podem surgir numa fase mais tardia.
As características faciais alteram-se consideravelmente com a idade. A esperança média de vida pode, devido às anomalias cardiovasculares e renais, estar ligeiramente diminuído, mas muitos estudos têm demonstrado a presença de excelente qualidade de vida em adultos com o síndrome de Williams. Apesar da estenose supravalvular aórtica e pulmonar, assim como a estenose da artéria renal e consequente hipertensão poderem causar problemas, nas crianças são mais os problemas comportamentais do que os do foro médico que preocupam os pais. Por um lado são indivíduos com personalidades sociais e de aspecto feliz, no entanto, a sua hiperactividade, labilidade emocional e ansiedade excessiva são particularidades que dificultam o seu relacionamento. A hiperacusia está frequentemente presente, podendo provocar reacções extremas a sons tais como o toque de campainhas e o ruído do cortador de relva, o que complica a terapêutica das alterações do comportamento. A microcefalia (cabeça pequena) e atraso do desenvolvimento são habituais e 59% destas crianças têm um QI inferior a 70. No entanto, muitas crianças cumprem os parâmetros apropriados à idade em diversas áreas tais como o reconhecimento visual, a linguagem expressiva e repetição de palavras.
Os mecanismos responsáveis pela hipercalcémia não estão completamente elucidados, podendo estar relacionados com uma resposta fisiológica diminuída a níveis normais de cálcio na alimentação e poderá contribuir para coarctação da aorta adquirida. Complicações mais tardias incluem o prolapso da válvula mitral, a úlcera péptica, a litíase biliar, a obesidade, as infecções urinárias e a diabetes mellitus. A hipercalcémia pode continuar a manifestar-se na vida adulta, por vezes resultando na excreção paradoxal da paratormona. Na 2ª e 3ª décadas a hipertensão arterial provocada pela calcificação dos rins e estenose das artérias renais torna-se mais frequente. Devido a estenoses das artérias cerebrais, alguns doentes sofrem acidentes vasculares cerebrais quando ainda jovens. Malformações do tracto urinário, incluindo divertículos vesicais podem surgir numa fase mais tardia.
Tratamento e Prevenção das Complicações
O
síndrome de Williams pode-se considerar uma doença multissistémica
progressiva. As medidas preventivas devem-se iniciar logo após o
diagnóstico com um estudo ecográfico para despiste de anomalias do
coração e rins. É necessário proceder à monitorização frequente da
hipertensão arterial, incluindo a avaliação da tensão arterial nos
quatro membros para exclusão de estenoses aórticas. Apesar dos problemas
alimentares, cólicas abdominais e obstipação poderem afectar o
crescimento, é razoável realizar uma abordagem conservadora uma vez que
as anomalias gastrointestinais são raras. A otite crónica exige
avaliações auditivas frequentes e quando necessário o envio para uma
consulta de otorrinolaringologia. O tratamento de problemas dentários
necessita da profilaxia da endocardite. Face às infecções urinárias
frequentes torna-se necessário avaliar a função renal periodicamente e
realizar um estudo ecográfico na infância e na adolescência.
Na adolescência, para além de se manter a vigilância dos sistemas já descritos, deve-se pesquisar a presença de uma curvatura exagerada da coluna vertebral (escoliose) e contracturas das articulações. Os problemas alimentares observados nos mais novos são ultrapassados, sendo a obesidade encontrada em 29% dos adultos. O comportamento e aproveitamento escolar, quando problemáticos carecem de medidas de apoio. A ansiedade pode estar associada à úlcera péptica e a litíase biliar é um diagnóstico possível em doentes com dores abdominais.
Na adolescência, para além de se manter a vigilância dos sistemas já descritos, deve-se pesquisar a presença de uma curvatura exagerada da coluna vertebral (escoliose) e contracturas das articulações. Os problemas alimentares observados nos mais novos são ultrapassados, sendo a obesidade encontrada em 29% dos adultos. O comportamento e aproveitamento escolar, quando problemáticos carecem de medidas de apoio. A ansiedade pode estar associada à úlcera péptica e a litíase biliar é um diagnóstico possível em doentes com dores abdominais.
Aconselhamento Genético
O
risco de recorrência para os filhos de pais saudáveis com crianças com
síndrome de Williams é inferior a 1%. Raramente encontram-se famílias
onde a hereditariedade é autossómica dominante. Face ao espectro de
problemas frequentes na criança com este síndrome, para além do apoio
das várias especialidades médicas, é também fundamental um apoio
psicossocial.
Para mais informações sobre o Síndrome de Williams podem consultar o seguinte site na internet:
www.williams-syndrome.org.uk
www.williams-syndrome.org.uk
Grupo de Apoio:
Associação Portuguesa de Portadores de Síndrome de Williams
Unidade de Desenvolvimento – Serviço de Pediatria,
Hospital de Santa Maria,
1695 Lisboa Codex
Associação Portuguesa de Portadores de Síndrome de Williams
Unidade de Desenvolvimento – Serviço de Pediatria,
Hospital de Santa Maria,
1695 Lisboa Codex
Síndrome de Angelman
Síndrome de Angelman


1. Pré-natal e história de nascimento normal com perímetro cefálico dentro da normalidade. 2. Ausência de defeitos sistêmicos de nascimento 3. Atraso de desenvolvimento evidente por volta dos 6 aos 12 meses4. Atraso do desenvolvimento neuropsicomotor (não há perda de aquisições)5. Exames laboratoriais normais6. Exames de imagem estrutural usando RM ou TC normais ou com anormalidades inespecíficas (atrofia cortical discreta ou desmielinização)** Estes achados são úteis como critérios de inclusão, entretanto caso não sejam observados, não devem excluir o diagnóstico.Quadro 4. Características Clínicas: Consistente (100%)Atraso de desenvolvimento, geralmente grave, alteração da linguagem, emissão de fonemas elementares e raramente articula palavras; comunicação receptiva e comunicação não-verbal maior do que verbal, distúrbios do movimento, usualmente ataxia à marcha e/ou tremores nos membros.
Comportamento único: sorrisos/ risadas freqüentes; comportamento alegre; facilmente excitável, com borboleteamento (movimento dos braços como em bater de asas); comportamento hipermotor (hiperatividade) e déficit de atençãoFreqüente (mais do que 80%)- Atraso no aumento do perímetro cefálico, resultando em microcefalia (absoluta ou relativa) por volta dos 2 anos de idade - Crises Epilépticas (instalação antes dos 3 anos de idade)- EEG anormal com padrões sugestivosAssociado (20% a 80%)Achatamento occipital
Depressão occipital
Protrusão lingual
Distúrbios de sucção e de deglutição
Problemas alimentares durante o período de lactente
Prognatismo
Boca ampla com dentes separados
Sialorréia
Comportamento oral-motor
Estrabismo
Hipopigmentação da pele, cabelos e íris (comparadas aos familiares, observada somente nos casos de deleção cromossômica)
Hiperreflexia
Posição elevada e semifletida dos braços, especialmente durante a deambulação
Hipersensibilidade ao calor
Distúrbios do sono
Atração ou fascínio pela águaIII. Características da Epilepsia:
A epilepsia ocorre em 80 a 90% dos pacientes com SA (Williams & Frias, 1982, Willems et al., 1987; Robb et al., 1989; Zori et al., 1992, Williams et al., 1995a). Na ASA já foram descritos todos os tipos de crises, parciais e generalizadas, com predomínio das generalizadas (Viani et al., 1995). Há uma prevalência elevada de crises febris, das ausências atípicas, das mioclonias e das tônico-clônica generalizadas. Desta forma, parece haver um padrão clínico, em relação aos tipos de crises mais freqüente nesta síndrome, o que poderia auxiliar na sua identificação e no seu diagnóstico. (Bower & Jeavons, 1967; Sugimoto et al., 1992; Matsumoto et al., 1992; Bourgeouis & Goutières, 1995; Viani et al., 1995; Dalla Bernardina et al., 1995; Guerrini et al., 1996; Laan et al., 1997a; Buoni et al., 1999)
As crises de ausência atípica são extremamente freqüentes na SA e caracterizam-se por perda total ou parcial do contato com o meio, queda do segmento cefálico e piscamento. Estas merecem destaque, pois freqüentemente apresentam-se como períodos de diminuição do contato com o meio e hipotonia com duração de dias a semanas (Sugimoto et al., 1992; Matsumoto et al., 1992; Laan et al., 1997a; Minassian et al., 1998), caracterizando o estado de mal de ausência atípica.
As crises de ausência atípica, assim como as crises mioclônicas, caracterizadas por abalos erráticos que acometem um ou vários segmentos corpóreos, são extremamente sutis podendo passar despercebidas, sendo reconhecidas através de estudos vídeo-eletrencefalográficos e poligráficos. A identificação e reconhecimento apropriado é essencial para a abordagem adequada com drogas anti-epilépticas.IV. Aspectos Eletrencefalográficos
Em idades extremas (lactente e idoso) o diagnóstico clínico precisa de considerações especiais devido aos aspectos evolutivos desta síndrome.1. Características Clínicas
No lactente: O fenótipo nos lactentes é sutil com anormalidades inespecíficas, o que torna o reconhecimento da síndrome muito difícil antes dos 3-4 anos de idade. Aos seis meses de vida aparecem as dificuldades alimentares e o atraso do desenvolvimento neuropsicomotor relacionado à hipotonia (Fryburg et al., 1991; Zori et al., 1992; Clayton-Smith, 1993; Buntinx et al., 1995). Por volta do primeiro aniversário, achados mais específicos se tornam evidentes como a microcefalia de instalação pós-natal, hipercinesia com movimentos em abalo e tremor, comportamento alegre com hiperatividade, crises epilépticas e distúrbios do sono (Zori et al., 1992).
A associação de hipopigmentação e distúrbios neurológicos (atraso de desenvolvimento e epilepsia) são pistas importantes para o diagnostico nesta faixa etária (Fryburg et al., 1991; Zori et al. 1992; Clayton-Smith, 1993; Buntinx et al., 1995). O quadro 5 ilustra as principais alterações descritas nos lactentes.
Na infância: As características clínicas da SA se tornam evidentes por volta dos 2o. ao 4o. ano de vida (Fryburg et al., 1991) persistindo pela infância e adolescência. Nesta fase para seu diagnóstico utiliza-se o Consenso para critérios Diagnósticos, anteriormente mencionado (Williams et al., 1995a).
No adulto: O adulto com SA pode apresentar quadro clínico caracterizado por traços faciais mais grosseiros (100%), escoliose torácica (71%), e pode haver restrição da marcha com uso de cadeiras de rodas (39%). Os episódios de riso são ainda observados no adulto (79%), porém com menor freqüência do que na criança (Laan et al., 1996). Não há limitações à longevidade, porém o idoso com SA pode apresentar maior incidência de problemas cardio-respiratórios do que na população em geral. Estes seriam decorrentes das contraturas e da cifoescoliose acentuada (Buntinx et al., 1995; Clayton-Smith, 1993; Sandanan et al., 1997).
2. Alterações da Epilepsia com a Idade
A idade de instalação ocorre, geralmente, na infância precoce, principalmente por volta dos 3 anos de idade (Sugimoto et al., 1992; Matsumoto et al., 1992; Dalla Bernardina et al., 1995; Casara et al., 1995; Viani et al., 1995; Minassian et al., 1998), embora a instalação mais tardia (por volta dos 5 anos) possa ocorrer (Laan et al., 1997a).
Parece haver, na maioria dos pacientes, um período de refratariedade e diversidade das crises epilépticas seguido da melhora ou controle das mesmas na infância tardia e adolescência (Clayton-Smith, 1993; Matsumoto et al., 1992; Viani et al., 1995).
3. Alterações do EEG com a Idade
A maior parte do conhecimento obtido sobre EEG vem de séries de pacientes pediátricos (Matsumoto et al., 1992; Sugimoto et al., 1992; Casara et al., 1995). Portanto, muito se sabe sobre o EEG na infância e muito pouco no adulto com SA.
Há, na maior parte das séries, assim como na nossa experiência pessoal, uma diminuição da freqüência dos padrões, que se tornam mais breves. Nesta fase, EEGs mais prolongados são necessários.VI. Correlação entre Fenótipo e Grupos Genéticos DistintosA maior parte dos estudos demonstra que pacientes sem deleção apresentam quadro clínico menos grave, o que facilitaria sua performance, entretanto dificultaria o seu diagnóstico (BOTTANI et al., 1994; BURGER et al., 1996; MONCLA et al., 1999b). Acredita-se que isto ocorra pela preservação de um maior contingente gênico que é a explicação mais provável e mais fácil, porém não a única.
Em relação à epilepsia, esta também parece ser mais grave nos pacientes com deleção do que nos demais grupos (Minassian et al.,1998; Valente et al, 2000).
O EEG, entretanto, parece ser um procedimento útil no auxílio ao diagnóstico, a despeito da ausencia de epilepsia ou de um fenótipo menos grave (Laan et al. 1997b; 1998b; Minassian et al., 1998; Moncla et al. 1999a; 199b).VII. DiagnósticoO diagnóstico da SA está, atualmente, embasado em uma tríade que engloba clínica, EEG e genética.
O diagnóstico da SA pode ser confirmado em 80% dos casos pelos estudos genéticos. Como o diagnóstico clínico é mais fácil entre 1 a 4 anos de idade (Fryburg et al., 1991; Magenis et al., 1990), as alterações eletrencefalográficas podem ser o primeiro sinal para uma avaliação diagnóstica (Boyd et al. 1988; 1997).
O EEG é particularmente importante para o diagnóstico em duas situações: nos pacientes com estudos genéticos negativos e no lactente.
O diagnóstico pré-natal da síndrome é possível com a análise do padrão de metilação do vilo coriônico ou amniocentese (CLAYTON-SMITH, 1991; Williams et al., 1995b).
As características clínicas recomendados seguem o Consenso para Critérios Diagnósticos (WILLIAMS et al., 1995a). Os exames genéticos para determinação da SA estão listados no quadro abaixo (Quadro 6).
IX. Tratamento
1. Terapia de ApoioSialorréia: após os 10 anos de idade, se não houver remissão espontânea, pode-se indicar um procedimento cirúrgico. O procedimento indicado foi descrito por VARMA et al. (1991), e conduzido com sucesso na última década (Henderson, 1993).Estrabismo: a correção do estrabismo inclui a avaliação oftalmológica, a correção de possíveis déficits visuais e o uso de tampões. Ajustes cirúrgicos podem ser indicados (Williams et al., 1995b).Albinismo e hipopigmentação: a hipersensibilidade ao sol torna importante a indicação de protetores solares (Williams et al., 1995b).Escoliose: afeta principalmente os adolescentes e adultos, com indicação cirúrgica (Sandanan et al., 1997).Comportamento: a orientação e a adequação dos pais pode resultar em melhora do desenvolvimento da criança (Summers et al., 1999). A modificação de comportamento pode levar alguns destes pacientes a ficarem independentes quanto à higiene diária e atividades da vida cotidiana, tais como a alimentação, vestir-se e atividades domésticas com alguma independência (Williams et al., 1995b).Linguagem: existem técnicas especiais de motivação para o desenvolvimento da linguagem não-verbal (linguagem através de figuras ou gestos) nos pacientes menos afetados (Williams et al., 1995b).Sono: o uso de sedativos tem sido postulado por alguns (Williams et al., 1995b).Contraturas: a fisioterapia deve ser encorajada para ajudar na mobilidade das articulações e prevenir a hipertonia. A terapia ocupacional e hidroterapia podem ser usadas no manejo dos pacientes com a síndrome.2. Tratamento das crises epilépticasLaan et al. (1997a) têm indicado o valproato como a droga de escolha no tratamento da SA, isolada ou em associação com benzodiazepínico, independente do tipo de crise epiléptica apresentada. O fenobarbital também parece ser efetivo no tratamento das crises epilépticas na SA.Estudo realizado no Reino Unido, através de um questionário enviado aos familiares, demonstrou que o valproato, o clonazepam e a lamotrigina como monoterapia ou em combinação foram as medicações mais eficazes no controle das crises e apresentaram menos efeitos colaterais no comportamento e nível de alerta do que a carbamazepina e a vigabatrina, que levaram a um aumento da freqüência e gravidade das crises, predominantemente mioclônicas e de ausência atípica (Kuenzle et al., 1998; PeruCca et al., 1998; Ruggieri & MCSHANE, 1998).As alternativas terapêuticas para o tratamento do estado de mal na SA são o ACTH (Matsumoto et al., 1992), corticosteróides (Boyd et al. 1988; Bourgeouis & Goutières, 1995) e valproato em monoterapia com altas doses ou associado ao clonazepam (Laan et al., 1997a). Outras opções que têm sido postulados para o tratamento da SA são o topiramato (FRANZ et al. 2001) e o piracetam (Guerrini et al., 1995; 1996; Genton et al.,1999; Obeso et al., 1988; Prazantelli & Nadi, 1996).
Dra. Kette Dualibi Ramos Valente MD, PhD
Laboratório de Neurofisiologia Clínica
Instituto de Psiquiatria - Hospital das Clínicas
Universidade de São Paulo
Dr. Ovídio Pires de Campos 785 - Cerqueira César
CEP 05403-010 - São Paulo/SPi

A primeira descrição de um paciente com tiques e comportamentos, que caracterizam a ST, ocorreu em 1825, pelo médico francês Jean Marc Gaspard Itard, que diagnosticou a maldição dos tiques na Marquesa de Dampierre (Itard, 1825). Entretanto, somente em 1884, esta patologia recebeu o nome de síndrome de Gilles de la Tourette (ST), quando o aluno Gilles de la Tourette, no Hospital de la Salpêtrière, relatou a patologia como um distúrbio caracterizado por tiques múltiplos, incluindo o uso involuntário ou inapropriado de palavras obscenas (coprolalia) e a repetição involuntária de um som, palavra ou frase de outrem (ecolalia), baseado nos relatos do próprio Itard (Gilles de la Tourette, 1885ab).
Nos últimos anos, a incidência de casos de ST vem crescendo em todo mundo, provavelmente, devido à maior disponibilidade de informações e conhecimento sobre esta doença pelas equipes de saúde que a diagnosticam (Hounie e Petribú, 1999).
O objetivo deste trabalho é abordar, de forma abrangente, diversos aspectos da ST, incluindo a sua definição, a etiologia, o quadro clínico, a epidemiologia, o diagnóstico e o tratamento, bem como, alguns de seus aspectos neurobiológicos e associações de apoio a portadores de ST.

Devido ao fato de a ST não apresentar um sintoma único, mas um conjunto de sinais e sintomas, a dificuldade no diagnóstico é evidente, quando se compara esta patologia com outras relacionadas, como: doença de Wilson, doença de Huntington, coréia de Sydenham, doença de Hallervorden-Spatz e com alguns tiques simples e múltiplos (Tabela 1). Dependendo da equipe de saúde que atende o paciente afetado pela ST, o seu diagnóstico pode demorar muito, sendo os sintomas atribuídos comumente a algum transtorno psiquiátrico. Esses diagnósticos demorados e/ou errôneos podem submeter os pacientes a tratamentos desnecessários e dispendiosos, antes de se definir propriamente a patologia e o procedimento correto a ser realizado.
No entanto, algumas características peculiares e o quadro clínico do paciente auxiliam no diagnóstico conclusivo da ST, onde sintomas como: a presença de múltiplos tiques motores e vocais, com início antes dos 18 anos de idade, sem origem em nenhuma resposta fisiológica (por exemplo, uso de estimulantes), com ocorrências diárias, estendendo-se por mais de um ano e com comprometimento social, ocupacional e/ou emocional, tornam-se decisivos para a definição do quadro patológico (Braunwald et al., 2002).
Estudos de tomografias de emissão têm revelado hipometabolismo e hipoperfusão em regiões do córtex frontal e temporal, no cíngulo, estriado e tálamo de pacientes com ST (George et al., 1992). Estes estudos de análise do metabolismo de glicose e do fluxo sangüíneo na região córtico-estriatal têm identificado anormalidades, principalmente envolvendo o gânglio de base e áreas corticais destes indivíduos. A observação de tomografia por emissão de pósitrons (PET) depois da injeção de [18F]2-fluoro-2-2desoxiglicose revelaram aumento bilateral simétrico ou diminuição da utilização da glicose dentro do gânglio de base e redução de atividade nos córtex frontal, cíngulo e insular (Baxter et al., 1990; Stoetter et al., 1992; Braun et al., 1995). Conjuntamente, estudos de fluxo sangüíneo cerebral, por tomografia de emissão de fóton (SPECT), têm identificado hipoperfusão do gânglio de base (Hall et al., 1991), sendo observada a redução do fluxo sangüíneo na região lenticular esquerda (Riddle et al., 1992; Moriarty et al., 1995). Estudos polissonográficos em pacientes com ST têm demonstrado, ainda, que estes apresentam distúrbios relacionados ao sono, apesar da tomografia cerebral computadorizada (TCC) geralmente apresentar-se sem anormalidades (Glaze et al., 1983; Robertson, 1989, Cohrs et al., 2001).
Do ponto de vista neuroquímico, diversas hipóteses sugerem o envolvimento do sistema dopaminérgico na patogênese da ST, visto que os neurolépticos, antagonistas da dopamina, são considerados efetivos no tratamento desta doença, por promover grande redução dos tiques (Singer et al., 1982; Golden, 1988; Shapiro et al., 1989; Singer, 1997). Por outro lado, os estimulantes como o metilfenidato, a cocaína, a pemolina e a L-dopa causam exarcebação dos tiques (Arzimanoglou, 1998). Com base nestes dados, a literatura sugere alguns mecanismos pelos quais o sistema dopaminérgico poderia estar envolvido, tais como, anormalidades na liberação de dopamina (Singer et al., 2002), hiperinervação dopaminérgica (Malison et al., 1995; Muller-Vahl et al., 2000) e a presença de receptores dopaminérgicos supersensíveis (Grice et al., 1996; Cruz et al., 1997; Díaz-Anzaldúa et al., 2004a) (Figura 1).
Anormalidades no reflexo de piscar os olhos presentes em pacientes com ST podem sugerir também aumento na atividade central dopaminérgica (Smith e Lees, 1989; Tulen et al., 1999; Raffaele et al., 2004). Além disso, um estudo recente, envolvendo análises pos-mortem de tecidos do córtex frontal e estriato, mostrou densidade elevada do receptor dopaminérgico D2 na região pré-frontal de pacientes com ST, o que reforça a importância do sistema dopaminérgico na patogênese da ST (Minzer et al., 2004).
O papel de outros neurotransmissores, tais como, acetilcolina, Gaba, sistema opióide endógeno, serotonina e norepinefrina (Brett et al., 1995; Comings, 1995; Comings et al., 1999; Hebebrand et al., 1997; Robertson, 2000; Eapen et al., 2001; Pauls, 2001; Eapen et al., 2004) vem sendo estudado, já que não se pode descartar o envolvimento de outros neurotransmissores dentro do circuito CSTC (Pauls, 2001). Devido ao fato de não se ter determinado, ainda, todos os fatores envolvidos no quadro neurológico anormal da ST, não se pode excluir a possibilidade da mesma ser uma síndrome multicausal.
Dentre as patologias associadas à ST, o transtorno de déficit de atenção, acompanhado da hiperatividade e de sintomas obsessivo-compulsivos, são descritos na maioria dos estudos relatados na literatura (21% a 90% e 50%, respectivamente) (Robertson e Yakely, 1996; Mercadante et al., 2004). Muitas das crianças afetadas pela ST e portadoras do transtorno de déficit de atenção e hiperatividade podem apresentar também deficiências de aprendizagem (Arzimanoglou, 1998).
*************************************************
A Síndrome de Angelman (S.A)
é um distúrbio neurológico que causa retardo mental, alterações do
comportamento e algumas características físicas distintivas. Ela foi
pela primeira vez relatada em 1965, quando um neurologista britânico,
Dr. Harry Angelman, descreveu 3 crianças com este quadro. Até 1987, o
interesse por esta doença foi bastante reduzido. Neste ano, observou-se
que a análise dos cromossomos de afetados por S.A. mostrava em cerca de
50% dos indivíduos a falta de uma pequena porção (deleção) do cromossomo
15. O que parecia ser uma situação muito rara mostrou-se bastante
frequente: estima-se atualmente que uma em cada quinze ou vinte mil
crianças são afetadas por esta doença.
I. Histórico
Em 1965 o pediatra britânico Harry Angelman,
descreveu três crianças, não-consangüíneas, com retardo mental que
apresentavam aparência similar, perfil comportamental peculiar
caracterizado por episódios de riso imotivado, movimentos em abalo
(robot-like) e crises epilépticas. A semelhança fenotípica apresentada
por estes pacientes com uma pintura de Giovanni Francesco Caroto
intitulada “Franciullo con su pupazzo”, vista pelo médico, anos antes na
cidade de Castelvecchio (Itália), levou o autor a nomeá-los “puppet
children” ou crianças marionetes.
Bower
& Jeavons, em 1967, ao descreverem dois casos similares aos
relatados por ANGELMAN (1965), enfatizaram o comportamento alegre dos
mesmos e sugeriram o termo “happy puppet syndrome”. Esta denominação
foi logo aceita e amplamente utilizada devido ao seu significado
evocativo, entretanto o termo “Síndrome de Angelman” (SA) foi adotado,
posteriormente, por não ter caráter depreciativo (Williams & Frias,
1982).
Seguiram-se
novos relatos de caso (Berg & Pakula, 1972; Mayo et al., 1973;
Moore & Jeavons, 1973; Elian, 1975; Dooley et al., 1981; Hersh et
al., 1981), com um número restrito de pacientes, que sugeriam uma doença
rara com natureza esporádica.
WilliamS
& Frias (1982) foram os primeiros a postular que a SA estivesse
provavelmente subdiagnosticada e subestimada e concluíram que havia
poucas evidências para uma herança mendeliana nesta síndrome.
O
relato de casos familiais (Kuroki et al., 1980; Pashayan et al., 1982;
Fisher et al., 1987) sugeria que na SA, até então considerada como
esporádica, havia mecanismos genéticos que precisavam ser revistos em
favor de um delineamento mais preciso.
Em
1987, o relato de uma deleção no braço longo do cromossomo 15
(15q11-q13) materno relacionada à SA (Kaplan et al., 1987; Magenis et
al., 1987), em contraposição a deleção 15q11-q13 paterna relacionada à
síndrome de Prader-Willi (SPW), permitiu reconhecer que as duas
síndromes tratar-se-iam de exemplos de imprinting genômico. Como
imprinting, entende-se a “expressão diferencial de alelos herdados
paterna ou maternalmente”, ou “expressão diferencial de alelos
dependente da origem parental da herança”.
Dentro
deste contexto, em 1988, Boyd et al. ao analisarem 19 pacientes,
definiram três padrões eletrencefalográficos sugestivos da SA, visando
auxiliar o diagnóstico, principalmente em idades precoces.
Em
1989, Robb et al. relataram as características clínicas da mais ampla
série até então publicada com 36 crianças com idades entre 1,6 e 13,5
anos, relacionando-as com a idade de aparecimento.
Posteriormente,
a publicação de outras séries na literatura com um número expressivo de
pacientes (Zori et al., 1992; Clayton-Smith, 1993; Williams et al.,
1995b) ampliou o conhecimento sobre as características clínicas da SA.
A
variabilidade clínica e genética demonstrada nestas publicações levou à
criação do “CONSENSO PARA CRITÉRIOS DIAGNÓSTICOS” (Williams et al.,
1995a).
Na
década de 90, outros mecanismos genéticos vieram a ser descritos
(dissomia uniparental paterna, mutação no centro de imprinting, mutação
no gene UBE3A), demonstrando tratar-se de uma síndrome determinada por
múltiplos mecanismos genéticos. Atualmente, a SA é um diagnóstico
clínico que pode ser confirmado pelos estudos genéticos em 80 a 85% dos
casos.
A atual prevalência estimada da SA é de 1/10000 a 1/20000 (PETERSEN et al., 1995).
Embora,
nos últimos 40 anos, muito se tenha avançado no reconhecimento clínico
desta síndrome, ainda se sabe pouco sobre as suas características
evolutivas e sobre a relação entre a real participação dos distintos
mecanismos genéticos na configuração do seu fenótipo.
ANGELMAN, 1965 – descrição de três crianças não consangüíneas com características semelhantes.
BOWERS
& JEAVONS, 1967 – descrição de mais dois casos com as mesmas
características e o uso do termo “happy puppet syndrome” é sugerido.
BERG & PAKULA, 1972 – descrição do 6o. caso. O termo “Angelman Syndrome” é usado pela primeira vez para nomear a síndrome.
MOORE & JEAVONS, 1973 – relato de mais dois casos, um cuja suspeita diagnóstica foi realizada através do eletrencefalograma.
BERGREEN,
1972; KIBEL & BURNESS, 1973; MASSEY & ROY, 1973 –relatos de
caso com ênfase para os aspectos clínicos da síndrome.
ELIAN, 1975 – revisão de 14 casos relatados em 10 anos após a primeira descrição. Ênfase sobre a raridade desta síndrome
Relatos de caso do final da década de 70: HALAL & CHAGNON, 1976; PELC et al., 1976.
II. Perfil Clínico
A
S.A não é uma síndrome homogênea, em relação às suas características
clínicas. Esta heterogeneidade parece estar relacionada a vários
fatores, entre eles a faixa etária (Magenis et al., 1990; VAN LIERDE et
al., 1990; Fryburg et al., 1991; BUNTINX et al., 1995; LAAN et al.,
1996; SANDANAM et al., 1997).
1. Critérios Diagnósticos
O
CONSENSO PARA CRITÉRIOS DIAGNÓSTICOS foi criado para definir as
características clínicas desta síndrome assim como a freqüência com que
ocorrem (Williams et al., 1995a) e estão representados nos quadros 3 e
4.
Quadro 3. História e Achados Laboratoriais** WILLIAMS et al., 1995
1. Pré-natal e história de nascimento normal com perímetro cefálico dentro da normalidade. 2. Ausência de defeitos sistêmicos de nascimento 3. Atraso de desenvolvimento evidente por volta dos 6 aos 12 meses4. Atraso do desenvolvimento neuropsicomotor (não há perda de aquisições)5. Exames laboratoriais normais6. Exames de imagem estrutural usando RM ou TC normais ou com anormalidades inespecíficas (atrofia cortical discreta ou desmielinização)** Estes achados são úteis como critérios de inclusão, entretanto caso não sejam observados, não devem excluir o diagnóstico.Quadro 4. Características Clínicas: Consistente (100%)Atraso de desenvolvimento, geralmente grave, alteração da linguagem, emissão de fonemas elementares e raramente articula palavras; comunicação receptiva e comunicação não-verbal maior do que verbal, distúrbios do movimento, usualmente ataxia à marcha e/ou tremores nos membros.
Comportamento único: sorrisos/ risadas freqüentes; comportamento alegre; facilmente excitável, com borboleteamento (movimento dos braços como em bater de asas); comportamento hipermotor (hiperatividade) e déficit de atençãoFreqüente (mais do que 80%)- Atraso no aumento do perímetro cefálico, resultando em microcefalia (absoluta ou relativa) por volta dos 2 anos de idade - Crises Epilépticas (instalação antes dos 3 anos de idade)- EEG anormal com padrões sugestivosAssociado (20% a 80%)Achatamento occipital
Depressão occipital
Protrusão lingual
Distúrbios de sucção e de deglutição
Problemas alimentares durante o período de lactente
Prognatismo
Boca ampla com dentes separados
Sialorréia
Comportamento oral-motor
Estrabismo
Hipopigmentação da pele, cabelos e íris (comparadas aos familiares, observada somente nos casos de deleção cromossômica)
Hiperreflexia
Posição elevada e semifletida dos braços, especialmente durante a deambulação
Hipersensibilidade ao calor
Distúrbios do sono
Atração ou fascínio pela águaIII. Características da Epilepsia:
A epilepsia ocorre em 80 a 90% dos pacientes com SA (Williams & Frias, 1982, Willems et al., 1987; Robb et al., 1989; Zori et al., 1992, Williams et al., 1995a). Na ASA já foram descritos todos os tipos de crises, parciais e generalizadas, com predomínio das generalizadas (Viani et al., 1995). Há uma prevalência elevada de crises febris, das ausências atípicas, das mioclonias e das tônico-clônica generalizadas. Desta forma, parece haver um padrão clínico, em relação aos tipos de crises mais freqüente nesta síndrome, o que poderia auxiliar na sua identificação e no seu diagnóstico. (Bower & Jeavons, 1967; Sugimoto et al., 1992; Matsumoto et al., 1992; Bourgeouis & Goutières, 1995; Viani et al., 1995; Dalla Bernardina et al., 1995; Guerrini et al., 1996; Laan et al., 1997a; Buoni et al., 1999)
As crises de ausência atípica são extremamente freqüentes na SA e caracterizam-se por perda total ou parcial do contato com o meio, queda do segmento cefálico e piscamento. Estas merecem destaque, pois freqüentemente apresentam-se como períodos de diminuição do contato com o meio e hipotonia com duração de dias a semanas (Sugimoto et al., 1992; Matsumoto et al., 1992; Laan et al., 1997a; Minassian et al., 1998), caracterizando o estado de mal de ausência atípica.
As crises de ausência atípica, assim como as crises mioclônicas, caracterizadas por abalos erráticos que acometem um ou vários segmentos corpóreos, são extremamente sutis podendo passar despercebidas, sendo reconhecidas através de estudos vídeo-eletrencefalográficos e poligráficos. A identificação e reconhecimento apropriado é essencial para a abordagem adequada com drogas anti-epilépticas.IV. Aspectos Eletrencefalográficos
Boyd et al. (1988) descreveram três padrões que caracterizam a SA e que devido à sua especificidade permitem o diagnóstico precoce.1. Padrões Eletrencefalográficos Sugestivos da SAOs três padrões eletrencefalográficos descritos caracterizam-se por:1) Surtos de atividade delta rítmico, de projeção generalizada, usualmente com predomínio frontal, de alta amplitude, com mais de 300µV com ou sem pequenas espículas superpostas;2) Surtos de atividade teta ritmada, com mais de 100 mV, com projeção generalizada ou nas regiões posteriores, não relacionada à sonolência e; 3) Surtos de ondas agudas ou ondas de 4 a 6 Hz de projeção nos quadrantes posteriores. Estes últimos são freqüentemente ativados pelo fechamento palpebral, sendo freqüentemente assimétricos e podendo ocasionalmente se espraiar para as regiões temporais.2. A possibilidade do diagnóstico precoceAs características do EEG sugestivas da SA não são vistas ao nascimento, mas aparecem gradativamente por volta dos 4 aos 9 meses (Boyd, 1997). O reconhecimento dos padrões eletrencefalográficos torna o diagnóstico mais fácil principalmente em idades mais precoces, mesmo antes que as crises tenham início ou que o fenótipo seja reconhecível (Boyd et al., 1988; Laan et al., 1997a).V. Variações do Fenótipo em Relação à Idade
Em idades extremas (lactente e idoso) o diagnóstico clínico precisa de considerações especiais devido aos aspectos evolutivos desta síndrome.1. Características Clínicas
No lactente: O fenótipo nos lactentes é sutil com anormalidades inespecíficas, o que torna o reconhecimento da síndrome muito difícil antes dos 3-4 anos de idade. Aos seis meses de vida aparecem as dificuldades alimentares e o atraso do desenvolvimento neuropsicomotor relacionado à hipotonia (Fryburg et al., 1991; Zori et al., 1992; Clayton-Smith, 1993; Buntinx et al., 1995). Por volta do primeiro aniversário, achados mais específicos se tornam evidentes como a microcefalia de instalação pós-natal, hipercinesia com movimentos em abalo e tremor, comportamento alegre com hiperatividade, crises epilépticas e distúrbios do sono (Zori et al., 1992).
A associação de hipopigmentação e distúrbios neurológicos (atraso de desenvolvimento e epilepsia) são pistas importantes para o diagnostico nesta faixa etária (Fryburg et al., 1991; Zori et al. 1992; Clayton-Smith, 1993; Buntinx et al., 1995). O quadro 5 ilustra as principais alterações descritas nos lactentes.
Quadro 5. Alterações mais freqüentes nos lactentes(BARAITSER
et al.,1987; ROBB et al., 1989; van LIERDE et al., 1990; FRYBURG et
al., 1991; ZORI et al., 1992; CLAYTON-SMITH et al., 1993): Manifestações Neurológicas:- Microcefalia com instalação pós-natal
- Atraso de desenvolvimento sem perda de aquisições
- Linguagem mínima ou ausente
- Crises epilépticas
- Hipotonia com hiperreflexia e hipercinesia
- Alteração de pigmentação (da hipopigmentação ao albinismo oculocutâneo)
- Manifestações oftalmológicas (hipoplasia macular, estrabismo e nistagmo)
- Anomalias craniofaciais são sutis, exceto por protrusão da língua e braquicefalia
- Distúrbios alimentares
- Comportamento alegre
- Atraso de desenvolvimento sem perda de aquisições
- Linguagem mínima ou ausente
- Crises epilépticas
- Hipotonia com hiperreflexia e hipercinesia
- Alteração de pigmentação (da hipopigmentação ao albinismo oculocutâneo)
- Manifestações oftalmológicas (hipoplasia macular, estrabismo e nistagmo)
- Anomalias craniofaciais são sutis, exceto por protrusão da língua e braquicefalia
- Distúrbios alimentares
- Comportamento alegre
Na infância: As características clínicas da SA se tornam evidentes por volta dos 2o. ao 4o. ano de vida (Fryburg et al., 1991) persistindo pela infância e adolescência. Nesta fase para seu diagnóstico utiliza-se o Consenso para critérios Diagnósticos, anteriormente mencionado (Williams et al., 1995a).
No adulto: O adulto com SA pode apresentar quadro clínico caracterizado por traços faciais mais grosseiros (100%), escoliose torácica (71%), e pode haver restrição da marcha com uso de cadeiras de rodas (39%). Os episódios de riso são ainda observados no adulto (79%), porém com menor freqüência do que na criança (Laan et al., 1996). Não há limitações à longevidade, porém o idoso com SA pode apresentar maior incidência de problemas cardio-respiratórios do que na população em geral. Estes seriam decorrentes das contraturas e da cifoescoliose acentuada (Buntinx et al., 1995; Clayton-Smith, 1993; Sandanan et al., 1997).
2. Alterações da Epilepsia com a Idade
A idade de instalação ocorre, geralmente, na infância precoce, principalmente por volta dos 3 anos de idade (Sugimoto et al., 1992; Matsumoto et al., 1992; Dalla Bernardina et al., 1995; Casara et al., 1995; Viani et al., 1995; Minassian et al., 1998), embora a instalação mais tardia (por volta dos 5 anos) possa ocorrer (Laan et al., 1997a).
Parece haver, na maioria dos pacientes, um período de refratariedade e diversidade das crises epilépticas seguido da melhora ou controle das mesmas na infância tardia e adolescência (Clayton-Smith, 1993; Matsumoto et al., 1992; Viani et al., 1995).
3. Alterações do EEG com a Idade
A maior parte do conhecimento obtido sobre EEG vem de séries de pacientes pediátricos (Matsumoto et al., 1992; Sugimoto et al., 1992; Casara et al., 1995). Portanto, muito se sabe sobre o EEG na infância e muito pouco no adulto com SA.
Há, na maior parte das séries, assim como na nossa experiência pessoal, uma diminuição da freqüência dos padrões, que se tornam mais breves. Nesta fase, EEGs mais prolongados são necessários.VI. Correlação entre Fenótipo e Grupos Genéticos DistintosA maior parte dos estudos demonstra que pacientes sem deleção apresentam quadro clínico menos grave, o que facilitaria sua performance, entretanto dificultaria o seu diagnóstico (BOTTANI et al., 1994; BURGER et al., 1996; MONCLA et al., 1999b). Acredita-se que isto ocorra pela preservação de um maior contingente gênico que é a explicação mais provável e mais fácil, porém não a única.
Em relação à epilepsia, esta também parece ser mais grave nos pacientes com deleção do que nos demais grupos (Minassian et al.,1998; Valente et al, 2000).
O EEG, entretanto, parece ser um procedimento útil no auxílio ao diagnóstico, a despeito da ausencia de epilepsia ou de um fenótipo menos grave (Laan et al. 1997b; 1998b; Minassian et al., 1998; Moncla et al. 1999a; 199b).VII. DiagnósticoO diagnóstico da SA está, atualmente, embasado em uma tríade que engloba clínica, EEG e genética.
O diagnóstico da SA pode ser confirmado em 80% dos casos pelos estudos genéticos. Como o diagnóstico clínico é mais fácil entre 1 a 4 anos de idade (Fryburg et al., 1991; Magenis et al., 1990), as alterações eletrencefalográficas podem ser o primeiro sinal para uma avaliação diagnóstica (Boyd et al. 1988; 1997).
O EEG é particularmente importante para o diagnóstico em duas situações: nos pacientes com estudos genéticos negativos e no lactente.
O diagnóstico pré-natal da síndrome é possível com a análise do padrão de metilação do vilo coriônico ou amniocentese (CLAYTON-SMITH, 1991; Williams et al., 1995b).
As características clínicas recomendados seguem o Consenso para Critérios Diagnósticos (WILLIAMS et al., 1995a). Os exames genéticos para determinação da SA estão listados no quadro abaixo (Quadro 6).
Quadro 6. Testes Genéticos da Síndrome de Angelman(WILLIAMS et al., 1995a; SMITH et al., 1995; 1997a; MALZAC et al., 1998)
A.
Estudo de alta resolução com banda G mostrando deleção do cr15q11-13.
Devido à possibilidade de falso-positivo e resultados negativos, este
teste não deve ser utilizado isoladamente e deve ser confirmado pelo
método de FISH, polimorfismo ou análise de metilação. B.
Fluorescence in situ hybridization (FISH) anormal indicando uma deleção
de seqüências do DNA clonadas do 15 q11-q13 que estão incluídas na
região de deleção da SA. O uso da sonda de FISH pericentromérica aumenta
a capacidade de detectar translocações sutis. C. Análise do
polimorfismo do DNA mostrando ausência de alelos maternos no loci 15
q11-q13, resultando ou de uma deleção do Cr materno ou de dissomia
uniparental. D. Análise do padrão de metilação do DNA (i.e. somente
padrão paterno) de seqüências de DNA clonadas do 15q11-q13
methylation-sensitive restriction endonucleases. Um padrão de metilação
anormal em um indivíduo sem 15q11-q13 não representa um teste padrão
isolado para o diagnóstico de DUP.E. Análise da mutação do UBE3A para a
detecção de mutações no E6-AP ubiqüitina-proteína ligase no Cr 15q11-13VIII. Diagnóstico DiferencialOutras
síndromes como a síndrome de Rett, a SPW, a encefalopatia crônica não
evolutiva, a síndrome de Lennox-Gastaut (Dulac & N’Guyen, 1993), o
autismo (FISHER, 1996) e a síndrome do retardo mental com talassemia
ligada ao Cr X (ATR-X) (WILKIE et al., 1990; Ogle et al., 1994) podem
mimetizar as características clinicas da SA, principalmente durante o
período de lactente, quando o EEG pode ser extremamente útil para o
diagnóstico diferencial (Laan et al., 1998a). Baseando-se,
exclusivamente, no EEG os principais diagnósticos diferenciais a serem
feitos são a síndrome de West, no lactente e a síndrome de
Lennox-Gastaut e síndrome de Doose, na infância (Matsumoto et al.,
1992). A síndrome do 4p- tem sido descrita como tendo características
eletrencefalográficas similares às da SA, apesar do quadro clínico
completamente distinto (S’GRO et al., 1995). O EEG é particularmente
útil no diagnóstico diferencial entre a SPW e a SA, em especial no
primeiro ano de vida quando várias características clínicas podem se
sobrepor como, por exemplo, a presença de albinismo oculocutâneo e
hipopigmentação (CREEL et al., 1986; KIRKILIONIS et al., 1991; SAITOH et
al., 1997; SPRITZ et al., 1997). O mesmo pode ocorrer nos estágios
iniciais da síndrome de Rett (LAAN et al., 1998a).
IX. Tratamento
1. Terapia de ApoioSialorréia: após os 10 anos de idade, se não houver remissão espontânea, pode-se indicar um procedimento cirúrgico. O procedimento indicado foi descrito por VARMA et al. (1991), e conduzido com sucesso na última década (Henderson, 1993).Estrabismo: a correção do estrabismo inclui a avaliação oftalmológica, a correção de possíveis déficits visuais e o uso de tampões. Ajustes cirúrgicos podem ser indicados (Williams et al., 1995b).Albinismo e hipopigmentação: a hipersensibilidade ao sol torna importante a indicação de protetores solares (Williams et al., 1995b).Escoliose: afeta principalmente os adolescentes e adultos, com indicação cirúrgica (Sandanan et al., 1997).Comportamento: a orientação e a adequação dos pais pode resultar em melhora do desenvolvimento da criança (Summers et al., 1999). A modificação de comportamento pode levar alguns destes pacientes a ficarem independentes quanto à higiene diária e atividades da vida cotidiana, tais como a alimentação, vestir-se e atividades domésticas com alguma independência (Williams et al., 1995b).Linguagem: existem técnicas especiais de motivação para o desenvolvimento da linguagem não-verbal (linguagem através de figuras ou gestos) nos pacientes menos afetados (Williams et al., 1995b).Sono: o uso de sedativos tem sido postulado por alguns (Williams et al., 1995b).Contraturas: a fisioterapia deve ser encorajada para ajudar na mobilidade das articulações e prevenir a hipertonia. A terapia ocupacional e hidroterapia podem ser usadas no manejo dos pacientes com a síndrome.2. Tratamento das crises epilépticasLaan et al. (1997a) têm indicado o valproato como a droga de escolha no tratamento da SA, isolada ou em associação com benzodiazepínico, independente do tipo de crise epiléptica apresentada. O fenobarbital também parece ser efetivo no tratamento das crises epilépticas na SA.Estudo realizado no Reino Unido, através de um questionário enviado aos familiares, demonstrou que o valproato, o clonazepam e a lamotrigina como monoterapia ou em combinação foram as medicações mais eficazes no controle das crises e apresentaram menos efeitos colaterais no comportamento e nível de alerta do que a carbamazepina e a vigabatrina, que levaram a um aumento da freqüência e gravidade das crises, predominantemente mioclônicas e de ausência atípica (Kuenzle et al., 1998; PeruCca et al., 1998; Ruggieri & MCSHANE, 1998).As alternativas terapêuticas para o tratamento do estado de mal na SA são o ACTH (Matsumoto et al., 1992), corticosteróides (Boyd et al. 1988; Bourgeouis & Goutières, 1995) e valproato em monoterapia com altas doses ou associado ao clonazepam (Laan et al., 1997a). Outras opções que têm sido postulados para o tratamento da SA são o topiramato (FRANZ et al. 2001) e o piracetam (Guerrini et al., 1995; 1996; Genton et al.,1999; Obeso et al., 1988; Prazantelli & Nadi, 1996).
Dra. Kette Dualibi Ramos Valente MD, PhD
Laboratório de Neurofisiologia Clínica
Instituto de Psiquiatria - Hospital das Clínicas
Universidade de São Paulo
Dr. Ovídio Pires de Campos 785 - Cerqueira César
CEP 05403-010 - São Paulo/SPi
n http://www.angelman.org.br/index.htm
Síndrome de Tourette
Síndrome de Tourette
A
síndrome de Tourette (ST) é uma patologia de comprometimento
psicossocial que acarreta alterações significativas na vida dos seus
portadores e respectivos familiares. Este artigo aborda diversos
aspectos relacionados a esta doença, incluindo etiologia, epidemiologia,
aspectos neurobiológicos, quadro clínico, diagnóstico, patologias
associadas e tratamento (clássico e alternativo). Neste trabalho, ainda
comparamos a ST com outras doenças, envolvendo tiques e mencionamos as
associações de apoio aos pacientes portadores de ST, que auxiliam no
tratamento e na socialização do paciente afetado.
A
síndrome de Tourette (ST) é uma patologia caracterizada pelo
comprometimento psicológico e social dos acometidos, causando impacto na
vida dos portadores e familiares (Hounie e Petribú, 1999). Ela é
geralmente associada ainda a uma variedade de problemas comportamentais e
emocionais (Singer e Minzer, 2003).
A primeira descrição de um paciente com tiques e comportamentos, que caracterizam a ST, ocorreu em 1825, pelo médico francês Jean Marc Gaspard Itard, que diagnosticou a maldição dos tiques na Marquesa de Dampierre (Itard, 1825). Entretanto, somente em 1884, esta patologia recebeu o nome de síndrome de Gilles de la Tourette (ST), quando o aluno Gilles de la Tourette, no Hospital de la Salpêtrière, relatou a patologia como um distúrbio caracterizado por tiques múltiplos, incluindo o uso involuntário ou inapropriado de palavras obscenas (coprolalia) e a repetição involuntária de um som, palavra ou frase de outrem (ecolalia), baseado nos relatos do próprio Itard (Gilles de la Tourette, 1885ab).
Nos últimos anos, a incidência de casos de ST vem crescendo em todo mundo, provavelmente, devido à maior disponibilidade de informações e conhecimento sobre esta doença pelas equipes de saúde que a diagnosticam (Hounie e Petribú, 1999).
O objetivo deste trabalho é abordar, de forma abrangente, diversos aspectos da ST, incluindo a sua definição, a etiologia, o quadro clínico, a epidemiologia, o diagnóstico e o tratamento, bem como, alguns de seus aspectos neurobiológicos e associações de apoio a portadores de ST.
A
ST, até pouco tempo, era considerada uma condição rara de índices de
baixíssima incidência na população mundial (0,5/1000, em 1984) (Bruun,
1984). Entretanto, tem-se observado, atualmente, através de estudos de
prevalência, o aumento de sua incidência nos últimos anos (Robertson e
Stern, 1998; Hounie e Petribú, 1999; Robertson, 2003; Eapen et al.,
2004).
Estudos atuais demonstram que a taxa de prevalência pode variar de 1% (Kadesjo e Gillberg, 2000) a 2,9% (Mason et al., 1998) em alguns grupos. Contudo, este dado deve ser subestimado, uma vez que depende, em parte, dos critérios e métodos utilizados e do tipo de estudo epidemiológico realizado (Robertson, 1989). Dados estatísticos internacionais mostram que esta síndrome é encontrada em vários países, independentemente de classe social ou de etnia, acometendo cerca de três a quatro vezes mais o sexo masculino, em relação ao sexo feminino (Robertson, 1989; Robertson, 1994; Arzimanoglou, 1998; Freeman et al., 2000; Robertson, 2000; Scahill et al., 2001).
Estudos mostram que a prevalência de ST é dez vezes maior em crianças e adolescentes (Burd et al., 1986), sendo que, quando tiques são considerados isoladamente, a freqüência aproximada varia de 1% a 13% nos meninos e 1% a 11% nas meninas (Leckman e Peterson, 1993). A razão para este aumento na detecção da incidência mundial da ST parece ser a melhoria na divulgação e no conhecimento das características clínicas da ST, entre os profissionais da área de saúde (Kushner, 1995; Hounie e Petribú, 1999).
Estudos atuais demonstram que a taxa de prevalência pode variar de 1% (Kadesjo e Gillberg, 2000) a 2,9% (Mason et al., 1998) em alguns grupos. Contudo, este dado deve ser subestimado, uma vez que depende, em parte, dos critérios e métodos utilizados e do tipo de estudo epidemiológico realizado (Robertson, 1989). Dados estatísticos internacionais mostram que esta síndrome é encontrada em vários países, independentemente de classe social ou de etnia, acometendo cerca de três a quatro vezes mais o sexo masculino, em relação ao sexo feminino (Robertson, 1989; Robertson, 1994; Arzimanoglou, 1998; Freeman et al., 2000; Robertson, 2000; Scahill et al., 2001).
Estudos mostram que a prevalência de ST é dez vezes maior em crianças e adolescentes (Burd et al., 1986), sendo que, quando tiques são considerados isoladamente, a freqüência aproximada varia de 1% a 13% nos meninos e 1% a 11% nas meninas (Leckman e Peterson, 1993). A razão para este aumento na detecção da incidência mundial da ST parece ser a melhoria na divulgação e no conhecimento das características clínicas da ST, entre os profissionais da área de saúde (Kushner, 1995; Hounie e Petribú, 1999).
A
ST é um distúrbio genético, de natureza neuropsiquiátrica,
caracterizado por fenômenos compulsivos, que, muitas vezes, resultam em
uma série repentina de múltiplos tiques motores e um ou mais tiques
vocais, durante pelo menos um ano, tendo início antes dos 18 anos de
idade (American Psychiatry Association, 1994; World Health Organization,
2000; Peterson, 2001; Pauls, 2003). Estes tiques podem ser
classificados como motores e vocais, subdividindo-se, ainda, em simples e
complexos. Geralmente, pacientes com ST apresentam, inicialmente,
tiques simples, evoluindo para os mais complexos; entretanto, o quadro
clínico pode variar de paciente para paciente (Leckman et al., 2001;
Mercadante et al., 2004).
Os
tiques motores classificam-se de acordo com o grupamento muscular
envolvido. Os tiques motores simples caracterizam-se por movimentos
abruptos, repetidos e sem propósito, envolvendo contrações de grupos
musculares funcionalmente relacionados (por exemplo, piscar os olhos e
movimentos de torção de nariz e boca). Os tiques motores complexos são
mais lentos, envolvem grupos musculares não relacionados funcionalmente e
parecem propositais (The Tourette Syndrome Classification Study Group,
1993; Hounie e Petribú, 1999). Os tiques motores complexos incluem
imitação de gestos realizados por outrem, sejam eles comuns (ecocinese)
ou obscenos (ecopraxia) e a realização de gestos obscenos (copropraxia)
(Braunwald et al., 2002). São freqüentemente observados compulsões e
gestos balizados, simétricos ou mesmo movimentos violentos com arremesso
de objetos.
Os
tiques vocais simples incluem, comumente, coçar a garganta e fungar,
enquanto que os tiques vocais complexos compreendem o uso involuntário
ou inapropriado de palavras chulas ou obscenas (coprolalia), repetição
de palavras ou frases (palilalia) e repetição involuntária das frases de
outrem (ecolalia) (Arzimanoglou, 1998; Hounie e Petribú, 1999).
Observa-se, também, o uso repetido de palavras aleatórias,
caracterizadas por sonoridade complexa ou exótica, arbitrariamente
colocadas entre ou no meio das frases. É importante ressaltar que a
simples presença do tique não caracteriza a ST, uma vez que, estudos
comprovaram que, 10% das crianças, presentes na população em geral,
apresentam tiques em algum momento. Todas as formas de tiques podem ser
exacerbadas por estresse, sendo normalmente reduzidas durante o sono e
em algumas atividades que exijam concentração (Arzimanoglou, 1998;
Mercadante et al., 2004). Estudos sobre tiques mostram que estes têm
início em torno dos 5 aos 10 anos de idade (Arzimanoglou, 1998) e
tornam-se mais pronunciados na faixa etária de 10 e 13 anos (Leckman et
al., 1998). Entretanto, cerca de 90% dos pacientes com ST apresentarão
remissão e mais de 40% estarão livres dos tiques até o fim da
adolescência (Arzimanoglou, 1998; Leckman et al., 1998; Burd et al.,
2001).
Com base nesses dados, o diagnóstico da ST é realizado através da presença de sinais e sintomas característicos e pela história de surgimento desses sintomas. Não existe, atualmente, nenhum teste laboratorial específico que confirme o diagnóstico da ST. Contudo, exames complementares (EEG, tomografia ou análises sangüíneas) podem ser úteis no diagnóstico diferencial da ST, contribuindo para a exclusão de outros distúrbios que possuem sintomas semelhantes (Jankovic, 2001).
Com base nesses dados, o diagnóstico da ST é realizado através da presença de sinais e sintomas característicos e pela história de surgimento desses sintomas. Não existe, atualmente, nenhum teste laboratorial específico que confirme o diagnóstico da ST. Contudo, exames complementares (EEG, tomografia ou análises sangüíneas) podem ser úteis no diagnóstico diferencial da ST, contribuindo para a exclusão de outros distúrbios que possuem sintomas semelhantes (Jankovic, 2001).
Devido ao fato de a ST não apresentar um sintoma único, mas um conjunto de sinais e sintomas, a dificuldade no diagnóstico é evidente, quando se compara esta patologia com outras relacionadas, como: doença de Wilson, doença de Huntington, coréia de Sydenham, doença de Hallervorden-Spatz e com alguns tiques simples e múltiplos (Tabela 1). Dependendo da equipe de saúde que atende o paciente afetado pela ST, o seu diagnóstico pode demorar muito, sendo os sintomas atribuídos comumente a algum transtorno psiquiátrico. Esses diagnósticos demorados e/ou errôneos podem submeter os pacientes a tratamentos desnecessários e dispendiosos, antes de se definir propriamente a patologia e o procedimento correto a ser realizado.
No entanto, algumas características peculiares e o quadro clínico do paciente auxiliam no diagnóstico conclusivo da ST, onde sintomas como: a presença de múltiplos tiques motores e vocais, com início antes dos 18 anos de idade, sem origem em nenhuma resposta fisiológica (por exemplo, uso de estimulantes), com ocorrências diárias, estendendo-se por mais de um ano e com comprometimento social, ocupacional e/ou emocional, tornam-se decisivos para a definição do quadro patológico (Braunwald et al., 2002).
Durante
a última década, tem sido possível observar significativo progresso na
investigação genética da etiologia da ST. Anormalidades cromossomiais em
indivíduos e famílias portadoras da ST têm sido estudadas, no intuito
de identificar genes como o gene A da monoamina-oxidase (MAOA) (Gade et
al., 1998; Díaz-Anzaldúa et al., 2004a) e regiões cromossômicas como a
18q22 (Cuker et al., 2004), 17q25 (Paschou et al., 2004) e 7q31
(Díaz-Anzaldúa et al., 2004b), que parecem estar envolvidas nesta
patologia (Brett et al., 1996; Kroisel et al,. 2001; Petek et al., 2001;
Crawford et al., 2003; State et al., 2003, Merette et al., 2000;
Simonic et al., 2001). Neste processo de identificação, evidências
sugerem que a ST seja um distúrbio genético de caráter autossômico
dominante, visto a freqüência de casos de tiques e manifestações
obsessivo-compulsivas entre familiares desses pacientes, observada em
estudos multicêntricos (Pauls et al., 1991; Eapen et al., 1993; Freeman
et al., 2000; Robertson, 2000). Até o presente momento, não foi possível
identificar um marcador genético de forma definitiva para a ST
(Díaz-Anzaldúa et al., 2004a).
Com isso, outros fatores também têm sido associados à patogênese da ST, tais como, o possível papel de infecções estreptocócicas na patogênese dos tiques (Cardona e Orefici, 2001; Hoekstra et al., 2004). Em alguns casos, as reinfecções por Streptococcus estão diretamente associadas com a recorrência de sintomas neuropsiquiátricos (Swedo et al., 1993 e 1998). A detecção de auto-anticorpos que reagem com o tecido cerebral em pacientes com tiques e/ou distúrbios obsessivo-compulsivos (Kiessling et al., 1993) levou os pesquisadores do Instituto Nacional de Doenças Mentais (National Institute of Mental Health – NIMH) a formularem critérios clínicos para um subgrupo de crianças com distúrbios obsessivo-compulsivo ou ST, nas quais as exacerbações dos sintomas são bruscas, dramáticas e temporariamente relacionadas com infecções Streptococcus ß-hemolítico do grupo A. Esse quadro clínico gerou a denominação distúrbios neuropsiquiátricos pediátricos auto-imune associados com infecções estreptocócicas (Swedo et al., 1997 e 1998). Anticorpos dirigidos contra a glicoproteína de oligodendrócito da mielina (MOG) também têm sido implicados como possível fator auto-imune na patogênese da ST (Huang et al., 2004). Existem ainda possíveis indicações do envolvimento de infecções não-estreptocócicas na etiologia da ST, como a relação temporal entre infecções respiratórias virais e exacerbação de tiques (Hoekstra et al., 2004) e o aumento de receptores para o fragmento Fc da imunoglobulina IgM, em linfócitos B, em pacientes portadores de tiques (Hoekstra et al., 2001). Entretanto estudos ainda estão sendo realizados para determinar a relação direta entre estes quadros infecciosos e a ST.
Eventos pré ou pós-parto, onde a gravidade dos estressores durante a gestação tem sido analisada como fatores que poderiam contribuir para o desenvolvimento de distúrbios de tiques e para a patogênese da ST (Hyde et al., 1992; Leckman et al., 1990; Robertson, 2000; Leckman e Herman, 2002). A análise de segregação de famílias indica que a ST é herdada de acordo com o padrão autossômico dominante com penetrância variável, dependendo do sexo (Eapen et al., 1993). Com isto, em virtude da elevada incidência de ST e tiques no sexo masculino, investiga-se a exposição do sistema nervoso central a altos níveis de testosterona e/ou outros hormônios gênero-específicos, como fatores importantes no desenvolvimento desta patologia (Leckman e Peterson, 1993, Eapen et al., 1993).


O estudo de imagens neurológicas tem possibilitado melhor entendimento sobre a base neural da ST, bem como, de sua provável patogênese (Gerard e Peterson, 2003). Diversas evidências do envolvimento do circuito córtico-estriato-tálamo-cortical (CSTC) e seus sistemas de neurotransmissão, associados com as características clínicas e comorbidades presentes na ST, têm sido amplamente divulgadas na literatura (Lou et al., 1989; Peterson, 2001; Singer e Wendlandt, 2001; Singer e Minzer, 2003). A supressão de tiques de pacientes submetidos a leucotomias e talamotomias, pela interrupção do circuito CSTC, apontam para o envolvimento direto deste na ST, sendo este fato observado através da visualização funcional da ressonância magnética, da análise das medidas de área do corpo caloso e pelo metabolismo da glicose e fluxo sangüíneo nas áreas corticais (Rauch et al., 1995; Singer e Minzer, 2003). Apesar de questionável (Chemali et al., 2003), a análise comparativa de imagens funcionais de ressonância magnética da atividade na região do córtex sensóriomotor e na área motora suplementar, durante a realização de uma tarefa motora predeterminada, em um grupo de pacientes com ST e um grupo controle, demonstraram ativação de regiões similares em ambos os grupos, mas com um número e dispersão das áreas de ativação da região do córtex sensório-motor apresentando-se visivelmente maiores em pacientes com ST (Figura 1) (Biswal et al., 1998). Estes resultados sugerem que, em tais pacientes, exista uma ativação anormal no córtex sensório-motor e nas áreas motoras suplementares. A visualização funcional utilizando a ressonância magnética nuclear, na qual foram comparadas imagens adquiridas, tanto em períodos de supressão voluntária de tiques, quanto durante a expressão espontânea, mostrou o efeito da supressão dos tiques na mudança de sinais nas regiões cortical e subcortical do cérebro. Isso indica que a supressão voluntária de tiques envolve a ativação do córtex préfrontal e do núcleo caudado, desativação bilateral do putâmen e do globo pálido (Peterson et al., 1998). As mudanças de atividade cortical e subcortical observadas nesse estudo sugerem, ainda, que a patogênese dos tiques envolve atividade neuronal dentro de circuitos neuronais subcorticais, reforçando, assim, a provável importância do circuito CSTC na fisiologia dos tiques e de seu controle voluntário (Peterson et al., 1998). Os resultados envolvendo imagens de ressonância magnética, obtidas durante a expressão de tiques fônicos, também sugerem que o circuito CSTC contribui, dentre outras coisas, para a inibição comportamental do indivíduo, pela modulação de atividade com o gânglio de base e o tálamo (Singer, 1997; Peterson et al.,1999; Gates et al., 2004). Isso poderia justificar a ocorrência de tiques em pacientes com ST, nos quais a atividade inibitória das projeções GABAérgicas do estriato para o globo pálido estivesse comprometida. No caso do corpo estriato não inibir a ação dos neurônios glutaminérgicos encontrados no tálamo, projeções excitatórias seriam então enviadas do tálamo para o córtex. Pacientes com ST são considerados incapazes de inibir o estímulo secundário ao fenômeno sensório-premonitório, que resulta na ativação do circuito motor e no desenvolvimento dos comportamentos motores e fônicos (Peterson et al., 1999). Anormalidades nos volumes dos gânglios de base no corpo caloso também foram observadas em portadores com ST (Peterson et al., 1993; Singer et al., 1993). A participação de uma alça associativa, contendo dois subcircuitos originários no córtex pré-central, pré-motor, parietal posterior e órbito-frontal lateral, com projeções para a cabeça do núcleo caudado, globo pálido medial e núcleos talâmicos anteriores e dorso medial, tem sido recentemente sugerida. O comprometimento desses circuitos estaria associado, dentre outros sintomas, à falha em responder às convenções sociais, com o uso de gestos e expressões inadequadas, fatos comumente presentes nos transtornos obsessivo-compulsivos (TOC) e na ST (Bar-Gad et al., 2003; Groenewegen, 2003).
Com isso, outros fatores também têm sido associados à patogênese da ST, tais como, o possível papel de infecções estreptocócicas na patogênese dos tiques (Cardona e Orefici, 2001; Hoekstra et al., 2004). Em alguns casos, as reinfecções por Streptococcus estão diretamente associadas com a recorrência de sintomas neuropsiquiátricos (Swedo et al., 1993 e 1998). A detecção de auto-anticorpos que reagem com o tecido cerebral em pacientes com tiques e/ou distúrbios obsessivo-compulsivos (Kiessling et al., 1993) levou os pesquisadores do Instituto Nacional de Doenças Mentais (National Institute of Mental Health – NIMH) a formularem critérios clínicos para um subgrupo de crianças com distúrbios obsessivo-compulsivo ou ST, nas quais as exacerbações dos sintomas são bruscas, dramáticas e temporariamente relacionadas com infecções Streptococcus ß-hemolítico do grupo A. Esse quadro clínico gerou a denominação distúrbios neuropsiquiátricos pediátricos auto-imune associados com infecções estreptocócicas (Swedo et al., 1997 e 1998). Anticorpos dirigidos contra a glicoproteína de oligodendrócito da mielina (MOG) também têm sido implicados como possível fator auto-imune na patogênese da ST (Huang et al., 2004). Existem ainda possíveis indicações do envolvimento de infecções não-estreptocócicas na etiologia da ST, como a relação temporal entre infecções respiratórias virais e exacerbação de tiques (Hoekstra et al., 2004) e o aumento de receptores para o fragmento Fc da imunoglobulina IgM, em linfócitos B, em pacientes portadores de tiques (Hoekstra et al., 2001). Entretanto estudos ainda estão sendo realizados para determinar a relação direta entre estes quadros infecciosos e a ST.
Eventos pré ou pós-parto, onde a gravidade dos estressores durante a gestação tem sido analisada como fatores que poderiam contribuir para o desenvolvimento de distúrbios de tiques e para a patogênese da ST (Hyde et al., 1992; Leckman et al., 1990; Robertson, 2000; Leckman e Herman, 2002). A análise de segregação de famílias indica que a ST é herdada de acordo com o padrão autossômico dominante com penetrância variável, dependendo do sexo (Eapen et al., 1993). Com isto, em virtude da elevada incidência de ST e tiques no sexo masculino, investiga-se a exposição do sistema nervoso central a altos níveis de testosterona e/ou outros hormônios gênero-específicos, como fatores importantes no desenvolvimento desta patologia (Leckman e Peterson, 1993, Eapen et al., 1993).
O estudo de imagens neurológicas tem possibilitado melhor entendimento sobre a base neural da ST, bem como, de sua provável patogênese (Gerard e Peterson, 2003). Diversas evidências do envolvimento do circuito córtico-estriato-tálamo-cortical (CSTC) e seus sistemas de neurotransmissão, associados com as características clínicas e comorbidades presentes na ST, têm sido amplamente divulgadas na literatura (Lou et al., 1989; Peterson, 2001; Singer e Wendlandt, 2001; Singer e Minzer, 2003). A supressão de tiques de pacientes submetidos a leucotomias e talamotomias, pela interrupção do circuito CSTC, apontam para o envolvimento direto deste na ST, sendo este fato observado através da visualização funcional da ressonância magnética, da análise das medidas de área do corpo caloso e pelo metabolismo da glicose e fluxo sangüíneo nas áreas corticais (Rauch et al., 1995; Singer e Minzer, 2003). Apesar de questionável (Chemali et al., 2003), a análise comparativa de imagens funcionais de ressonância magnética da atividade na região do córtex sensóriomotor e na área motora suplementar, durante a realização de uma tarefa motora predeterminada, em um grupo de pacientes com ST e um grupo controle, demonstraram ativação de regiões similares em ambos os grupos, mas com um número e dispersão das áreas de ativação da região do córtex sensório-motor apresentando-se visivelmente maiores em pacientes com ST (Figura 1) (Biswal et al., 1998). Estes resultados sugerem que, em tais pacientes, exista uma ativação anormal no córtex sensório-motor e nas áreas motoras suplementares. A visualização funcional utilizando a ressonância magnética nuclear, na qual foram comparadas imagens adquiridas, tanto em períodos de supressão voluntária de tiques, quanto durante a expressão espontânea, mostrou o efeito da supressão dos tiques na mudança de sinais nas regiões cortical e subcortical do cérebro. Isso indica que a supressão voluntária de tiques envolve a ativação do córtex préfrontal e do núcleo caudado, desativação bilateral do putâmen e do globo pálido (Peterson et al., 1998). As mudanças de atividade cortical e subcortical observadas nesse estudo sugerem, ainda, que a patogênese dos tiques envolve atividade neuronal dentro de circuitos neuronais subcorticais, reforçando, assim, a provável importância do circuito CSTC na fisiologia dos tiques e de seu controle voluntário (Peterson et al., 1998). Os resultados envolvendo imagens de ressonância magnética, obtidas durante a expressão de tiques fônicos, também sugerem que o circuito CSTC contribui, dentre outras coisas, para a inibição comportamental do indivíduo, pela modulação de atividade com o gânglio de base e o tálamo (Singer, 1997; Peterson et al.,1999; Gates et al., 2004). Isso poderia justificar a ocorrência de tiques em pacientes com ST, nos quais a atividade inibitória das projeções GABAérgicas do estriato para o globo pálido estivesse comprometida. No caso do corpo estriato não inibir a ação dos neurônios glutaminérgicos encontrados no tálamo, projeções excitatórias seriam então enviadas do tálamo para o córtex. Pacientes com ST são considerados incapazes de inibir o estímulo secundário ao fenômeno sensório-premonitório, que resulta na ativação do circuito motor e no desenvolvimento dos comportamentos motores e fônicos (Peterson et al., 1999). Anormalidades nos volumes dos gânglios de base no corpo caloso também foram observadas em portadores com ST (Peterson et al., 1993; Singer et al., 1993). A participação de uma alça associativa, contendo dois subcircuitos originários no córtex pré-central, pré-motor, parietal posterior e órbito-frontal lateral, com projeções para a cabeça do núcleo caudado, globo pálido medial e núcleos talâmicos anteriores e dorso medial, tem sido recentemente sugerida. O comprometimento desses circuitos estaria associado, dentre outros sintomas, à falha em responder às convenções sociais, com o uso de gestos e expressões inadequadas, fatos comumente presentes nos transtornos obsessivo-compulsivos (TOC) e na ST (Bar-Gad et al., 2003; Groenewegen, 2003).
Estudos de tomografias de emissão têm revelado hipometabolismo e hipoperfusão em regiões do córtex frontal e temporal, no cíngulo, estriado e tálamo de pacientes com ST (George et al., 1992). Estes estudos de análise do metabolismo de glicose e do fluxo sangüíneo na região córtico-estriatal têm identificado anormalidades, principalmente envolvendo o gânglio de base e áreas corticais destes indivíduos. A observação de tomografia por emissão de pósitrons (PET) depois da injeção de [18F]2-fluoro-2-2desoxiglicose revelaram aumento bilateral simétrico ou diminuição da utilização da glicose dentro do gânglio de base e redução de atividade nos córtex frontal, cíngulo e insular (Baxter et al., 1990; Stoetter et al., 1992; Braun et al., 1995). Conjuntamente, estudos de fluxo sangüíneo cerebral, por tomografia de emissão de fóton (SPECT), têm identificado hipoperfusão do gânglio de base (Hall et al., 1991), sendo observada a redução do fluxo sangüíneo na região lenticular esquerda (Riddle et al., 1992; Moriarty et al., 1995). Estudos polissonográficos em pacientes com ST têm demonstrado, ainda, que estes apresentam distúrbios relacionados ao sono, apesar da tomografia cerebral computadorizada (TCC) geralmente apresentar-se sem anormalidades (Glaze et al., 1983; Robertson, 1989, Cohrs et al., 2001).
Do ponto de vista neuroquímico, diversas hipóteses sugerem o envolvimento do sistema dopaminérgico na patogênese da ST, visto que os neurolépticos, antagonistas da dopamina, são considerados efetivos no tratamento desta doença, por promover grande redução dos tiques (Singer et al., 1982; Golden, 1988; Shapiro et al., 1989; Singer, 1997). Por outro lado, os estimulantes como o metilfenidato, a cocaína, a pemolina e a L-dopa causam exarcebação dos tiques (Arzimanoglou, 1998). Com base nestes dados, a literatura sugere alguns mecanismos pelos quais o sistema dopaminérgico poderia estar envolvido, tais como, anormalidades na liberação de dopamina (Singer et al., 2002), hiperinervação dopaminérgica (Malison et al., 1995; Muller-Vahl et al., 2000) e a presença de receptores dopaminérgicos supersensíveis (Grice et al., 1996; Cruz et al., 1997; Díaz-Anzaldúa et al., 2004a) (Figura 1).
Anormalidades no reflexo de piscar os olhos presentes em pacientes com ST podem sugerir também aumento na atividade central dopaminérgica (Smith e Lees, 1989; Tulen et al., 1999; Raffaele et al., 2004). Além disso, um estudo recente, envolvendo análises pos-mortem de tecidos do córtex frontal e estriato, mostrou densidade elevada do receptor dopaminérgico D2 na região pré-frontal de pacientes com ST, o que reforça a importância do sistema dopaminérgico na patogênese da ST (Minzer et al., 2004).
O papel de outros neurotransmissores, tais como, acetilcolina, Gaba, sistema opióide endógeno, serotonina e norepinefrina (Brett et al., 1995; Comings, 1995; Comings et al., 1999; Hebebrand et al., 1997; Robertson, 2000; Eapen et al., 2001; Pauls, 2001; Eapen et al., 2004) vem sendo estudado, já que não se pode descartar o envolvimento de outros neurotransmissores dentro do circuito CSTC (Pauls, 2001). Devido ao fato de não se ter determinado, ainda, todos os fatores envolvidos no quadro neurológico anormal da ST, não se pode excluir a possibilidade da mesma ser uma síndrome multicausal.
Dentre as patologias associadas à ST, o transtorno de déficit de atenção, acompanhado da hiperatividade e de sintomas obsessivo-compulsivos, são descritos na maioria dos estudos relatados na literatura (21% a 90% e 50%, respectivamente) (Robertson e Yakely, 1996; Mercadante et al., 2004). Muitas das crianças afetadas pela ST e portadoras do transtorno de déficit de atenção e hiperatividade podem apresentar também deficiências de aprendizagem (Arzimanoglou, 1998).
Uma
vez diagnosticada a ST em um indivíduo, aspectos diretamente
relacionados aos sintomas, como a localização, freqüência, intensidade,
complexidade e interferência na vida diária dos pacientes, devem ser
cuidadosamente avaliados antes de se iniciar qualquer conduta
terapêutica. O diagnóstico e tratamento precoces são essenciais, a fim
de reduzir ou evitar possíveis conseqüências psicológicas para o
paciente. A escolha do tipo de tratamento deve ser apropriada para cada
portador da ST, podendo incluir abordagens farmacológica e a
psicológica. Esta última, além do tratamento psicoterápico do paciente,
orienta pais, familiares e pessoas próximas ao mesmo, sobre as
características da doença e o modo de lidar com o indivíduo afetado
(Hounie e Petribú, 1999; Mercadante et al., 2004).

A natureza intencional dos tiques permite uma abordagem terapêutica comportamental com o objetivo de reduzir sua freqüência através da interrupção da seqüência estímulo-resposta. A reversão de hábito temse mostrado eficaz para o tratamento dos tiques na ST (Wilhelm et al., 2003; Verdellen et al., 2004). Desse modo, um programa terapêutico multidisciplinar deve ser estabelecido, em colaboração com familiares e o paciente, visando o apoio psicológico e a reintegração social do mesmo (Arzimanoglou, 1998).
O uso de medicamentos ou outras técnicas podem trazer tanto benefícios quanto efeitos colaterais e, portanto, a abordagem farmacológica deve ser considerada somente quando os benefícios da intervenção forem superiores aos efeitos adversos. Além disso, fatores psicológicos e sociais podem influenciar na evolução da resposta terapêutica em pacientes com ST (Sandor, 2003).
Até o momento a ST não tem cura, sendo que o tratamento farmacológico é utilizado para o alívio e controle dos sintomas apresentados. O medicamento é administrado em pequenas doses, com aumentos graduais até que se atinja o máximo de supressão dos sintomas com o mínimo de efeitos colaterais. A posologia dos medicamentos varia para cada paciente, necessitando ser avaliada atentamente pelo médico (Associação Brasileira de Síndrome de Tourette, Tiques e Transtorno Obsessivo-compulsivo, 2004).
No grupo dos medicamentos utilizados no tratamento dos portadores de transtornos de tiques, encontram-se os antidepressivos tricíclicos, usados também no transtorno de déficit de atenção e hiperatividade associados, onde é contra-indicado o uso de psicoestimulantes (Spencer et al., 1994). Estudos mostram que os antagonistas dos receptores dopaminérgicos reduzem a freqüência e a severidade dos tiques em cerca de 70% dos casos (Shapiro e Shapiro, 1998). Essas observações sugerem que o bloqueio dos receptores dopaminérgicos tipo 2 é o ponto central para a eficácia do tratamento (Scahill et al., 2000; Sandor, 2003) e por isso, os antagonistas dos receptores de dopamina são largamente utilizados (Hounie e Petribú, 1999).
O haloperidol, um neuroléptico com ação antagônica sobre os receptores dopaminérgicos, iniciou a era do tratamento farmacológico da ST, há cerca de 40 anos (Caprini, 1961; Seignot, 1961; Sandor, 2003). Este medicamento é utilizado inicialmente em pequenas doses (0,25 a 0,5 mg ao dia) com aumentos de 0,5 mg por semana até o máximo de 2 a 3 mg ao dia. A dose deve ser individualizada para cada paciente, havendo relatos entre 0,5 a 40 mg ao dia. Para o controle dos tiques, este medicamento é requerido em baixas doses na maioria dos pacientes com ST, como indicado por um recente estudo, no qual uma dose equivalente à cerca de 2 mg de haloperidol, por dia, atingia a saturação de cerca de 80% dos receptores dopaminérgicos tipo 2. Este nível de efeito seria essencial para a eficácia do tratamento e a diminuição dos tiques (Fitzgerald et al., 2000). O tratamento, entretanto, apresenta uma série de efeitos adversos, como sintomas extrapiramidais de características parkinsonianas, sedação, disforia, hiperfagia com ganho de peso e, o mais grave, discinesia tardia (Sallee et al., 1997; Hounie e Petribú, 1999).
A pimozida tem sido proposta como alternativa ao haloperidol, devido à eficácia comparável e menor ocorrência de efeitos adversos extrapiramidais. Por outro lado, este medicamento possui efeitos de maior gravidade, envolvendo o sistema cardiovascular, incluindo ainda sedação e disfunção cognitiva (Ross e Moldofsky, 1978; Shapiro et al., 1989). A pimozida vem sendo utilizada largamente para o tratamento de ST em doses que variam de 1 a 20 mg ao dia (Sallee et al., 1997; Hounie e Petribú, 1999). Embora raro em baixas doses, o uso prolongado de pimozida pode causar o prolongamento da onda QT (Fulop et al., 1987), e portanto, recomenda-se realizar o exame eletrocardiográfico (ECG), durante a terapia de manutenção (Scahill et al., 2000).
Na prática clínica, existe uma forte tendência a substituir os chamados antagonistas típicos de receptores dopaminérgicos, tais como, haloperidol e pimozida, pelos antagonistas atípicos (risperidona, olanzapina e, em menor extensão, quetiapina) (Van der Linden et al., 1994; Brunn e Budman, 1996; Sandor, 2003) ou por agonistas dos receptores alfa-2-adrenérgicos como a clonidina e a guanfacina (Gaffney et al., 2002). Os antagonistas atípicos oferecem poucos efeitos adversos e vêm substituindo, gradualmente, o haloperidol e a pimozida, como suporte principal no tratamento dos tiques (Sandor, 2003).
Novas opções de tratamento parecem incluir a risperidona, um neuroléptico atípico com potentes propriedades antagonistas D2 e 5-HT2. A propriedade antagonista 5-HT2 parece conferir proteção contra efeitos colaterais extrapiramidais, talvez diminuindo a incidência de discenesia tardia (Scahill et al., 2000). Os efeitos adversos do uso da risperidona são a sedação, aumento de apetite e elevação dos níveis de prolactina. Embora sintomas extrapiramidais sejam eventualmente observados, esses são muito menos freqüentes do que com o uso de haloperidol ou pimozida. Disforia e depressão podem ocorrer em indivíduos predispostos durante o tratamento com risperidona (Margolese et al., 2002). Alguns estudos indicam efeito terapêutico da quetiapina no tratamento de tiques, sendo requerida em altas doses (200 a 500 mg ao dia), entretanto, pode provocar o aumento de peso corporal do paciente (Chanob et al., 2001).
A olanzapina surge como uma droga eficaz no tratamento da ST. Estudos mais recentes mostram que 50% dos pacientes, em uso de olanzapina por oito semanas, apresentaram redução global dos tiques graves e 75% apresentaram melhora parcial do quadro, bem como, de sintomas agressivos presentes na síndrome, sem efeitos colaterais significativos, se comparados com os apresentados no uso dos neurolépticos típicos (Budman et al., 2001; Stamenkovic et al., 2000; Stephens et al., 2004).
Moléculas que mimetizam agonistas alfa-adrenérgicos, como a clonidina e a guanfacina, também podem ser utilizadas no tratamento da ST, apresentando resultados positivos em estudos controle (Peterson et al., 1999). A clonidina é uma droga não-neuroléptica que tem sido usada no tratamento da ST desde a década de 1970 (Cohen et al., 1979). Ela age como um agonista pré-sináptico dos receptores alfa-2, deprimindo o sistema noradrenérgico e promovendo a redução e a freqüência dos tiques (Scahill et al., 2000; Tourette’s Syndrome Study Group, 2002; Sandor, 2003). Por ser um agente anti-hipertensivo, sua administração deve ser acompanhada de monitoramento da pressão arterial. A suspensão abrupta do medicamento tem sido associada a efeito rebote, nos valores de pressão arterial (Leckman et al., 1986). Um estudo comparativo entre o uso da clonidina e da risperidona em crianças e adolescentes, portadores da ST, mostrou equivalência terapêutica entre as duas substâncias reduzindo, respectivamente em 26% e 36% os tiques e em 33% e 66% os sintomas obsessivocompulsivos (Gaffney et al., 2002).
Outro agonista seletivo para o receptor alfa-2 é a guanfacina que tem sido sugerida como substituta à clonidina no tratamento da ST (Sandor, 2003; Mercadante et al., 2004). Esta troca pode trazer benefícios, principalmente quando o transtorno de déficit de atenção e hiperatividade fazem parte do quadro apresentado pelo paciente (Chappell et al., 1995; Scahill et al., 2001).
Outras drogas tais como a nicotina, tetrabenazina e benzodiazepina também são atualmente empregadas em tratamentos alternativos da ST, bem como, a injeção de toxina botulínica, que pode ser uma boa opção terapêutica para o tratamento de tiques motores e em alguns casos para tiques vocais (Sweet et al., 1974; Jankovic e Orman, 1988; Marras et al., 2001; Silver et al., 2001; Mercadante et al., 2004).
Finalmente, resultados positivos foram verificados com o uso da tiaprida, da sulpirida e da amisulprida (Fountoulakis et al., 2004), com melhoras substanciais dos sintomas. A primeira mostrou-se eficaz no controle dos tiques em crianças, com a vantagem de não produzir efeitos discinéticos marcantes (Eggers et al., 1988).
A natureza intencional dos tiques permite uma abordagem terapêutica comportamental com o objetivo de reduzir sua freqüência através da interrupção da seqüência estímulo-resposta. A reversão de hábito temse mostrado eficaz para o tratamento dos tiques na ST (Wilhelm et al., 2003; Verdellen et al., 2004). Desse modo, um programa terapêutico multidisciplinar deve ser estabelecido, em colaboração com familiares e o paciente, visando o apoio psicológico e a reintegração social do mesmo (Arzimanoglou, 1998).
O uso de medicamentos ou outras técnicas podem trazer tanto benefícios quanto efeitos colaterais e, portanto, a abordagem farmacológica deve ser considerada somente quando os benefícios da intervenção forem superiores aos efeitos adversos. Além disso, fatores psicológicos e sociais podem influenciar na evolução da resposta terapêutica em pacientes com ST (Sandor, 2003).
Até o momento a ST não tem cura, sendo que o tratamento farmacológico é utilizado para o alívio e controle dos sintomas apresentados. O medicamento é administrado em pequenas doses, com aumentos graduais até que se atinja o máximo de supressão dos sintomas com o mínimo de efeitos colaterais. A posologia dos medicamentos varia para cada paciente, necessitando ser avaliada atentamente pelo médico (Associação Brasileira de Síndrome de Tourette, Tiques e Transtorno Obsessivo-compulsivo, 2004).
No grupo dos medicamentos utilizados no tratamento dos portadores de transtornos de tiques, encontram-se os antidepressivos tricíclicos, usados também no transtorno de déficit de atenção e hiperatividade associados, onde é contra-indicado o uso de psicoestimulantes (Spencer et al., 1994). Estudos mostram que os antagonistas dos receptores dopaminérgicos reduzem a freqüência e a severidade dos tiques em cerca de 70% dos casos (Shapiro e Shapiro, 1998). Essas observações sugerem que o bloqueio dos receptores dopaminérgicos tipo 2 é o ponto central para a eficácia do tratamento (Scahill et al., 2000; Sandor, 2003) e por isso, os antagonistas dos receptores de dopamina são largamente utilizados (Hounie e Petribú, 1999).
O haloperidol, um neuroléptico com ação antagônica sobre os receptores dopaminérgicos, iniciou a era do tratamento farmacológico da ST, há cerca de 40 anos (Caprini, 1961; Seignot, 1961; Sandor, 2003). Este medicamento é utilizado inicialmente em pequenas doses (0,25 a 0,5 mg ao dia) com aumentos de 0,5 mg por semana até o máximo de 2 a 3 mg ao dia. A dose deve ser individualizada para cada paciente, havendo relatos entre 0,5 a 40 mg ao dia. Para o controle dos tiques, este medicamento é requerido em baixas doses na maioria dos pacientes com ST, como indicado por um recente estudo, no qual uma dose equivalente à cerca de 2 mg de haloperidol, por dia, atingia a saturação de cerca de 80% dos receptores dopaminérgicos tipo 2. Este nível de efeito seria essencial para a eficácia do tratamento e a diminuição dos tiques (Fitzgerald et al., 2000). O tratamento, entretanto, apresenta uma série de efeitos adversos, como sintomas extrapiramidais de características parkinsonianas, sedação, disforia, hiperfagia com ganho de peso e, o mais grave, discinesia tardia (Sallee et al., 1997; Hounie e Petribú, 1999).
A pimozida tem sido proposta como alternativa ao haloperidol, devido à eficácia comparável e menor ocorrência de efeitos adversos extrapiramidais. Por outro lado, este medicamento possui efeitos de maior gravidade, envolvendo o sistema cardiovascular, incluindo ainda sedação e disfunção cognitiva (Ross e Moldofsky, 1978; Shapiro et al., 1989). A pimozida vem sendo utilizada largamente para o tratamento de ST em doses que variam de 1 a 20 mg ao dia (Sallee et al., 1997; Hounie e Petribú, 1999). Embora raro em baixas doses, o uso prolongado de pimozida pode causar o prolongamento da onda QT (Fulop et al., 1987), e portanto, recomenda-se realizar o exame eletrocardiográfico (ECG), durante a terapia de manutenção (Scahill et al., 2000).
Na prática clínica, existe uma forte tendência a substituir os chamados antagonistas típicos de receptores dopaminérgicos, tais como, haloperidol e pimozida, pelos antagonistas atípicos (risperidona, olanzapina e, em menor extensão, quetiapina) (Van der Linden et al., 1994; Brunn e Budman, 1996; Sandor, 2003) ou por agonistas dos receptores alfa-2-adrenérgicos como a clonidina e a guanfacina (Gaffney et al., 2002). Os antagonistas atípicos oferecem poucos efeitos adversos e vêm substituindo, gradualmente, o haloperidol e a pimozida, como suporte principal no tratamento dos tiques (Sandor, 2003).
Novas opções de tratamento parecem incluir a risperidona, um neuroléptico atípico com potentes propriedades antagonistas D2 e 5-HT2. A propriedade antagonista 5-HT2 parece conferir proteção contra efeitos colaterais extrapiramidais, talvez diminuindo a incidência de discenesia tardia (Scahill et al., 2000). Os efeitos adversos do uso da risperidona são a sedação, aumento de apetite e elevação dos níveis de prolactina. Embora sintomas extrapiramidais sejam eventualmente observados, esses são muito menos freqüentes do que com o uso de haloperidol ou pimozida. Disforia e depressão podem ocorrer em indivíduos predispostos durante o tratamento com risperidona (Margolese et al., 2002). Alguns estudos indicam efeito terapêutico da quetiapina no tratamento de tiques, sendo requerida em altas doses (200 a 500 mg ao dia), entretanto, pode provocar o aumento de peso corporal do paciente (Chanob et al., 2001).
A olanzapina surge como uma droga eficaz no tratamento da ST. Estudos mais recentes mostram que 50% dos pacientes, em uso de olanzapina por oito semanas, apresentaram redução global dos tiques graves e 75% apresentaram melhora parcial do quadro, bem como, de sintomas agressivos presentes na síndrome, sem efeitos colaterais significativos, se comparados com os apresentados no uso dos neurolépticos típicos (Budman et al., 2001; Stamenkovic et al., 2000; Stephens et al., 2004).
Moléculas que mimetizam agonistas alfa-adrenérgicos, como a clonidina e a guanfacina, também podem ser utilizadas no tratamento da ST, apresentando resultados positivos em estudos controle (Peterson et al., 1999). A clonidina é uma droga não-neuroléptica que tem sido usada no tratamento da ST desde a década de 1970 (Cohen et al., 1979). Ela age como um agonista pré-sináptico dos receptores alfa-2, deprimindo o sistema noradrenérgico e promovendo a redução e a freqüência dos tiques (Scahill et al., 2000; Tourette’s Syndrome Study Group, 2002; Sandor, 2003). Por ser um agente anti-hipertensivo, sua administração deve ser acompanhada de monitoramento da pressão arterial. A suspensão abrupta do medicamento tem sido associada a efeito rebote, nos valores de pressão arterial (Leckman et al., 1986). Um estudo comparativo entre o uso da clonidina e da risperidona em crianças e adolescentes, portadores da ST, mostrou equivalência terapêutica entre as duas substâncias reduzindo, respectivamente em 26% e 36% os tiques e em 33% e 66% os sintomas obsessivocompulsivos (Gaffney et al., 2002).
Outro agonista seletivo para o receptor alfa-2 é a guanfacina que tem sido sugerida como substituta à clonidina no tratamento da ST (Sandor, 2003; Mercadante et al., 2004). Esta troca pode trazer benefícios, principalmente quando o transtorno de déficit de atenção e hiperatividade fazem parte do quadro apresentado pelo paciente (Chappell et al., 1995; Scahill et al., 2001).
Outras drogas tais como a nicotina, tetrabenazina e benzodiazepina também são atualmente empregadas em tratamentos alternativos da ST, bem como, a injeção de toxina botulínica, que pode ser uma boa opção terapêutica para o tratamento de tiques motores e em alguns casos para tiques vocais (Sweet et al., 1974; Jankovic e Orman, 1988; Marras et al., 2001; Silver et al., 2001; Mercadante et al., 2004).
Finalmente, resultados positivos foram verificados com o uso da tiaprida, da sulpirida e da amisulprida (Fountoulakis et al., 2004), com melhoras substanciais dos sintomas. A primeira mostrou-se eficaz no controle dos tiques em crianças, com a vantagem de não produzir efeitos discinéticos marcantes (Eggers et al., 1988).
A
Associação da Síndrome de Tourette é uma entidade norte-americana,
fundada na década de 1970, que reúne pacientes com ST e seus familiares.
Esta associação desenvolve materiais educacionais destinados a
professores e se dedica ao desenvolvimento de pesquisas e divulgação
científica sobre ST, financia pesquisas, promove congressos e reúne os
maiores pesquisadores mundiais da área. Além disso, mobiliza milhões de
dólares por ano para pesquisas e tratamento da ST (Wertheim, 1981;
Tourette Syndrome Association , 2004).
Em 1996, foi criada no Brasil a Associação de Pacientes com Síndrome de Tourette, Tiques e Transtorno Obsessivo-compulsivo (ASTOC), que tem como objetivos divulgar e informar médicos e pacientes sobre a patologia, arrecadar doações para a formação de um fundo de pesquisa, que atenda às necessidades e interesses dos portadores da síndrome, incluindo a área de socialização dos pacientes, que é amplamente assistida pela associação.
A ASTOC, localizada em São Paulo, já tem núcleos em formação em outros estados do país (Associação Brasileira de Síndrome de Tourette, Tiques e Transtorno Obsessivo-compulsivo, 2004).
Em 1996, foi criada no Brasil a Associação de Pacientes com Síndrome de Tourette, Tiques e Transtorno Obsessivo-compulsivo (ASTOC), que tem como objetivos divulgar e informar médicos e pacientes sobre a patologia, arrecadar doações para a formação de um fundo de pesquisa, que atenda às necessidades e interesses dos portadores da síndrome, incluindo a área de socialização dos pacientes, que é amplamente assistida pela associação.
A ASTOC, localizada em São Paulo, já tem núcleos em formação em outros estados do país (Associação Brasileira de Síndrome de Tourette, Tiques e Transtorno Obsessivo-compulsivo, 2004).
Um
bom exemplo do quanto a ST atinge os seus portadores é o depoimento de
um paciente por ela acometido: “...Ter a Tourette é uma loucura, é como
estar bêbado o tempo todo. Estar sob o efeito do haloperidol é monótono,
faz a gente ficar sóbrio e quadrado, e nenhum dos dois estados é
realmente livre. Vocês, ‘normais’, que têm os transmissores certos nos
lugares certos, nas horas certas, em seus cérebros, têm todos os
sentimentos, todos os estilos disponíveis o tempo todo – seriedade,
leviandade, o que for adequado. Nós que temos a Tourette, não, somos
forçados à leviandade pela síndrome e forçados à seriedade quando
tomamos o remédio. Vocês são livres, têm um equilíbrio natural: nós
temos de nos contentar com um equilíbrio artificial...” O relato deste
paciente, enquadrado no terceiro grupo da ST e atualmente assistido pelo
neurologista Oliver Sacks, é um exemplo do quanto esta síndrome
complexa compromete a vida de seu portador (Sacks, 1985). Este paciente
tem sua vida controlada, não só pelo medicamento, mas também pela
síndrome, cujo mecanismo e tratamento ainda não estão amplamente
conhecidos. Ainda é necessária uma melhor compreensão dos aspectos
biológicos, genéticos e comportamentais da ST, incluindo sua vasta
divulgação na sociedade em geral, e não somente na comunidade médica, de
forma a facilitar o diagnóstico, o tratamento e o prognóstico de
pacientes com ST (Pauls, 2003; Eapen et al., 2004).
in http://www.hcnet.usp.br/
Síndrome de Asperger
Síndrome de Asperger

Como consequência destas dificuldades os portadores de Síndrome de Asperger acabam por se isolar e limitar os seus interesses a determinados temas assuntos, atitude que prejudica ainda mais a sua relação com o outro. Calcula-se que em Portugal existam cerca de 40.000 portadores de Síndrome de Asperger afectando maioritariamente os rapazes.
O Diagnóstico precoce é essencial para proporcionar aos portadores, os recursos necessários e a que têm direito que lhes permitam atingir o seu potencial, o qual muitas vezes é extraordinário, como pessoas verdadeiramente integradas na sociedade.

Origem
A primeira pesquisa neste campo foi realizada pelo psicólogo austríaco, Hans Asperger, em 1943 – mas apenas nos últimos 20 anos aumentou a informação disponível e o conhecimento da classe médica e do público em geral sobre o que se entendeu chamar Síndroma de Asperger. Para isto muito contribui o trabalho de especialistas como Tony Attwood, Lorna Wing, entre outros. O Síndroma de Asperger foi oficialmente classificado nos EUA como uma disfunção psicológica em 1994 que afecta o modo como uma pessoa comunica e se relaciona com os outros. Entre outras características das crianças/jovens com este Síndroma, podemos destacar as seguintes:

- Dificuldade na comunicação;
- Dificuldade no relacionamento social;
- Dificuldade no pensamento abstracto.
As pessoas com síndrome de Asperger têm problemas de linguagem em menor escala do que as classificadas como autistas, falam mais fluentemente e não têm dificuldades de aprendizagem tão marcadas. Têm normalmente inteligência (Q.I.) média ou mesmo acima da média e muitas não são diagnosticadas como tal, sendo muitas vezes referidas, pela família e professores, como estranhos, excêntricos, originais, diferentes, extravagantes ou esquisitos. Os casos menos pronunciados, podem fazer excelentes progressos, ter sucesso, e mesmo continuar os estudos ao nível universitário e arranjar um emprego.
Interacção Social
Ao contrário dos autistas “clássicos”, que normalmente estão ausentes e desinteressados do mundo que os rodeia, muitas crianças com síndroma de Asperger querem ser sociáveis e gostam do contacto humano. Têm no entanto dificuldade em perceber sinais não-verbais, incluindo os sentimentos traduzidos em expressões faciais, o que levanta problemas em criar e manter relações com pessoas que não percebem esta dificuldade. Precisam de aprender aspectos do convívio social que nós adquirimos sem pensar, como a entoação da voz, linguagem corporal e expressões faciais. Essencialmente, a criança com síndroma de Asperger:
- Isola-se socialmente, mas pode não se preocupar com isso;
- Pode ficar tensa e agitada ao tentar lidar com as abordagens e as exigências sociais de terceiros;
- Começa a ter consciência de que os seus colegas têm amizades, sobretudo quando atinge a adolescência. Nessa altura, pode querer ter os seus amigos, mas não tem nenhuma estratégia para desenvolver e consolidar amizades;
- Tem dificuldade em seguir as “deixas” sociais;
- Pode ter um comportamento socialmente inaceitável.

Comunicação em contextos sociais
- Pode ter uma linguagem aparentemente perfeita, mas com tendência para ser formal e pedante. “Como está? Chamo-me João” pode ser uma saudação típica de um adolescente com síndroma de Asperger – mas é precisamente isso que o distancia dos seus colegas, expondo-o ao ridículo.
- Tem frequentemente uma voz sem expressão (monocórdica). Também pode ter dificuldade em interpretar as diferentes entoações de terceiros. Quase toda a gente sabe dizer se uma pessoa está zangada, aborrecida ou radiante apenas pela entoação (ou inflexão de voz). Muitas vezes, a criança com síndroma de Asperger não consegue ter este tipo de discernimento, o que pode originar algumas situações complicadas. Por exemplo, um professor teve de combinar um sinal visual com um aluno – “Quando eu tirar os óculos enquanto estiver a olhar para ti, já sabes que estou zangado contigo”. Levantar a voz não tinha nenhum efeito sobre a criança.
- Também pode ter dificuldade em utilizar e interpretar comunicação não verbal como, por exemplo, linguagem corporal, gestos e expressões faciais.
- Pode compreender os outros de forma muito literal. Um exemplo: após o Rui se ter recusado terminantemente a ajudar a avó a arrumar a cozinha, esta disse-lhe: “Deves pensar que tens o rei na barriga!” O Rui respondeu-lhe: “Na barriga? Não é possível engolir um rei!”, não percebendo que a avó se referia à sua teimosia em não a ajudar. A criança não reagiria a uma expressão como “Está calor aqui” – enquanto todas as outras pessoas perceberiam de imediato o sinal para abrir uma janela.
Pensamento abstracto
Podem ser excelentes na memorização de factos e números mas têm normalmente dificuldade ao nível do pensamento abstracto. Isto é causa frequente de problemas na aprendizagem, em ambiente escolar, de matérias como o português ou filosofia. No entanto, podem ser excelentes a matemática ou geografia.
Interesses especiais
As crianças com síndroma de Asperger desenvolvem interesses obsessivos e podem em consequência disto adquirir um conhecimento enciclopédico sobre determinada matéria. Podem ficar fascinados com horários de comboios, um certo programa de televisão ou previsões meteorológicas. Isto pode originar-lhes alguma frustração por não entenderem que os seus interesses não são partilhados pelos outros. No entanto, estas obsessões podem ser aproveitadas, e conduzir a boas oportunidades profissionais e de investigação.
Gosto por Rotinas
Não gostam de alterações ou mudanças. Podem impor as suas rotinas, tais como insistir em seguir sempre o mesmo caminho para a escola. Na escola podem ficar nervosos com uma alteração no horário, ou mudança de professor. Gostam normalmente de ter uma rotina diária coerente e imutável. Se trabalham de acordo com um horário, um atraso inesperado, devido a um demora nos transportes ou a problemas de tráfego, podem torná-los muito nervosos ou ansiosos.
Sensibilidade Sensorial
Têm uma resposta exagerada a alguns estímulos, podendo ser afectados quaisquer dos 5 sentidos. Por exemplo, podem ter vómitos com um determinado sabor ou cheiro, podem não suportar que lhe mexam na cabeça, podem não gostar de tocar em determinadas texturas, podem não suportar determinados sons por demasiado intensos ou agudos.
Coordenação Motora
São normalmente desajeitados e têm dificuldade na coordenação motora, (atar os sapatos, andar de bicicleta, etc.). A sua forma de andar é peculiar. Essencialmente, a criança/jovem com tal síndroma:

- Pode ter movimentos bruscos e desastrados;
- Tem frequentemente problemas de organização – não consegue orientar-se nem reunir o material de que precisa;
- Tem dificuldade em escrever e desenhar ordenadamente e, muitas vezes, não termina as tarefas.
Vulnerabilidades
As crianças e adultos com síndroma de Asperger são muito vulneráveis. A adolescência amplifica a sua luta interna, gerando grande ansiedade. A possibilidade de suicídio deve ser uma preocupação. Problemas que podem surgir englobam, entre outros: vulnerabilidade ao abuso, (praxe, gozo, exploração, etc), porque os seus comportamentos são vistos como excêntricos ou peculiares, têm dificuldade de adaptação e encontram-se sozinhos; deficiente visão de conjunto devido ao seu interesse no detalhe (em consequência, não conseguem antever os resultados das suas acções ou palavras, nem colocar os assuntos em contexto); grande ansiedade quando há alterações da rotina do dia-a-dia, (p. exp., atraso de um autocarro, mudança de professor, desaparecimento da marca favorita, etc.); dificuldade em planear porque isso requer a capacidade de pensar em hipóteses e prever consequências; dificuldade em expressar os seus pensamentos, em comunicar os seus medos, os seus problemas e frustrações.
O que causa a Síndroma de Asperger?

Actualmente não existem em Portugal instituições dedicadas exclusivamente às crianças com síndroma de Asperger. Algumas andam nas escolas do ensino regular, onde o seu progresso depende do ambiente gerado à sua volta e do apoio e encorajamento de pais e professores. Outras frequentam Unidades de Intervenção Especializada, vocacionadas para crianças com problemas mais graves de desenvolvimento. As crianças/jovens com esta síndroma são mais vulneráveis porque, por um lado, podem não ter sido devidamente diagnosticadas, por outro, porque os seus problemas de aprendizagem são menos óbvios do que os de outras crianças. São por isso, normalmente, um alvo preferencial do abuso físico e verbal por parte dos seus colegas, o que os pode tornar especialmente frustrados ou angustiados. Ao crescerem tomam melhor consciência da sua diferença e podem ter tendência para a solidão e depressão. Normalmente querem ser sociáveis mas têm dificuldade em criar e manter amizades, mas o futuro não necessita de ser obrigatoriamente negro. Em adultos podem ter grande sucesso nas carreiras que escolhem, potenciando as suas qualidades de obstinação, memória e facilidade para a matemática, e podem desenvolver amizades duradouras. Como trabalhadores estes indivíduos têm características muito prezadas – pontualidade, fiabilidade e dedicação – no entanto é essencial que o ambiente de trabalho que o rodeia seja harmonioso e compreenda às suas características.
INTERVENÇÃO PEDAGÓGICA
O papel do(s) professor(s) da turma é central para a educação da criança/jovem com síndroma de Asperger. Cabe-lhe(s) assegurar que todos os alunos da turma são educados a um nível que seja adequado às necessidades de cada um. Para isso, é necessário criar um ambiente que encoraje a valorização dos indivíduos e reconheça os diferentes estilos de aprendizagem inerentes a cada um. Esse ambiente baseia-se na compreensão das necessidades de todos os alunos incluindo as crianças/jovens com síndroma de Asperger. As áreas específicas sobre as quais o professor da turma se deve debruçar são:
- Criação de um ambiente de trabalho calmo;
- Garantia de que a estrutura da sala de aulas está perfeitamente definida;
- Modificação das tarefas para tirar partido e consolidar as forças da criança;
- Garantia de que a criança compreende o que se espera dela;
- Introdução gradual da escolha, encorajando a tomada de decisões;
- Estratificação das tarefas, aumentando gradualmente as exigências feitas à criança;
- Orientação da atenção da criança a nível individual, em vez de se basear em instruções dadas a toda a turma;
- Acesso à formação disponível; Planeamento de Adequações Curriculares;
- Registo e monitorização dos progressos;
- Avaliação de estratégias de intervenção;
- Trabalho em estreita relação com a rede de apoio disponível;
- Estabelecimento e manutenção de ligações casa/escola.
Embora possa parecer inicialmente um conjunto de expectativas estimulante, na realidade não passa de um simples exemplo de práticas recomendadas na sala de aula. Um pensamento importante a não esquecer é que a criança com síndroma de Asperger faz parte do todo da comunidade escolar e deve ser aceite e apoiada por toda a comunidade escolar. Muitas vezes, estas crianças sentem-se mais à vontade a desempenhar tarefas de aprendizagem do que a brincar. Pode ser fácil lidar com a rotina e a estrutura explícita de uma tarefa de aprendizagem, desde que não seja essencial a interacção social com outras crianças. Devido à natureza subtil e penetrante da limitação, é na banalidade da existência momento a momento que podem ocorrer dificuldades para a criança com síndroma de Asperger. Deste modo, o professor tem de estar preparado para actuar a um nível igualmente subtil e penetrante, para estar presente na construção do dia da criança, para fazer parte do ambiente “protésico”, para fazer de tradutor, intermediário e, sempre que possível:
- Compreender a capacidade limitada da criança para interpretar as “deixas” sociais;
- Interpretar situações para a criança;
- Mostrar à criança o que se espera dela;
- Ajudar e ensinar as capacidades adequadas de interacção social como, por exemplo, revezar-se;
- Orientar os colegas no modo de interagir com o aluno, solicitando a sua ajuda;
- Compreender as dificuldades subtis da linguagem e da comunicação;
- Prestar atenção (“ouvir”) o padrão de utilização da linguagem da criança e estar alerta para as dificuldades de interpretação;
- Explicar, mostrar e esclarecer a criança em caso de confusão;
- Ajudar a criança a desenvolver urna utilização adequada da linguagem e a desenvolver uma consciência;
- Compreender as origens da rigidez e obsessão no comportamento da criança;
- Antecipar o que irá causar ansiedade e fazer as alterações necessárias;
- Analisar e simplificar situações ou actividades que causem alarme;
- Tornar os procedimentos associados a tarefas visualmente explícitos, através de estímulos visuais e pictóricos;
- Prestar apoio à criança nas actividades físicas, caso haja problemas de destreza;
- Ajudar a simplificar as tarefas de redacção;
- Estar preparado para os períodos de ansiedade com actividades adequadas de redução de tensão (stress);
- Avaliar regularmente o potencial para aumentar a autonomia da criança;
- Identificar as falhas no desenvolvimento de capacidades de autoajuda em áreas como vestir-se e lavar-se. Integrá-las no programa da criança;
- Identificar qualquer dificuldade de organização e produzir auxiliares práticos e visuais para ajudar a criança;
- Prestar apoio na ligação casa/escola e registar os progressos;
- Propor estratégias adequadas de recompensa nas Adequações Curriculares;
- Saber quando e como pedir ajuda ao docente de educação especial;
- Agir como elo de ligação regular com o docente de educação especial;
- Desenvolver e manter sistemas úteis de monitorização, avaliação e registo.
Além disso, o professor tem de ser:
- Calmo;
- Positivo;
- Coerente.
Principais dicas para os professores utilizarem com as crianças com síndroma de Asperger:
Comunicação
- Simplifique a linguagem utilizada.
- Dê uma instrução de cada vez, em vez de uma série de instruções.
- Mantenha as expressões faciais e os gestos simples e explícitos.
- Dê tempo à criança para responder. Utilize auxiliares visuais adicionais para ajudar a criança a compreender.
- Seja sensível às tentativas da criança para comunicar.
- Prepare situações que encorajem a criança a tentar comunicar.
Interacção social
- Compreenda que a criança pode sentir-se ameaçada pela proximidade extrema de terceiros – sobretudo de outras crianças da mesma idade.
- Permita que a criança se isole.
- Acompanhe o ritmo da criança ao tentar desenvolver uma interacção – talvez seja necessário “regredir” em termos de desenvolvimento.
- Identifique as preferências e as antipatias das crianças a nível social – utilize este conhecimento ao planear as actividades.
- É mais provável que a criança interaja com pessoas conhecidas, por isso dê-lhe tempo para ficar a conhecê-lo e não a confunda com muitas mudanças de pessoal.
Comportamento
- Adopte uma abordagem com a máxima coerência.
- Ajude a criança a compreender o que se espera dela através de rotinas explícitas e previsíveis.
- Introduza a mais pequena alteração de forma gradual.
- Ajude a explicar as mudanças através de auxiliares visuais.
- Se a criança ficar agitada, compreenda que as estratégias habituais para acalmar uma criança (por exemplo, tentar sentá-la junto a si) podem ter o efeito oposto e ela acabar por ficar ainda mais agitada.
- Se a criança tiver uma obsessão, não tente detê-la. Com o tempo, talvez consiga limitá-la – entretanto, utilize-a de forma positiva.
Em geral
- Os resultados e os progressos podem ser lentos – não desista! (Muitas vezes, construir uma relação demora muito tempo.)
- Cada criança é única – o que resulta para uma pode não resultar para outra.
- Cada criança é instável – por isso, se ela estiver num “dia não”, não pense que a culpa é sua.
- Se tudo o resto falhar, deixe-a sozinha. Amanhã é outro dia!
As palavras-chave da intervenção são rotina, clareza e coerência. As pequenas mudanças no ambiente da sala de aulas, que simplifiquem a organização e a estrutura da sala e das tarefas, podem ajudar a criança a perceber quais são as expectativas. É possível fazer alterações úteis nas seguintes áreas do ambiente:
- Ambiente físico e sensorial;
- Ambiente de linguagem e comunicação;
- Ambiente social;
- Ambiente curricular.
Em seguida, é possível integrar intervenções específicas que permitam à criança desenvolver capacidades e compreensão nas áreas de Interacção Social, Comunicação Social e Imaginação e Jogo Social. Por outras palavras, é necessário que a abordagem seja compensatória, mas também terapêutica.
Ambiente físico e sensorial
Um ambiente congestionado, imprevisível e em constante mudança só vai confundir a criança com síndroma de Asperger e deixá-la ansiosa. Através da análise do ambiente de aprendizagem da criança, é possível imaginar as modificações que poderão ajudá-la.
Organização e estrutura
A maioria das salas de aula é organizada do ponto de vista social. O papel da compreensão social é assumido pelos professores que podem, efectivamente, não ter consciência desse papel na organização da sala de aula, por exemplo, no trabalho em grupo, na partilha de material, em ouvir uma instrução enquanto grupo. Imensas salas de aula da J. Infância e 1.º ciclo são organizadas por grupos, sendo que os grupos se movem pela sala de aulas para aceder às tarefas. Poderá ser necessário permitir que a criança tenha o seu próprio “espaço” algumas vezes durante o dia. Isto é particularmente importante sempre que é introduzida uma nova tarefa, uma vez que a tensão de fazer parte de um grupo pode limitar a compreensão. Este “espaço” não tem de ser um posto de trabalho individual sofisticado – uma carteira junto à mesa do grupo, de costas para as distracções, pode ser extremamente eficaz. É útil se a criança ocupar habitualmente um determinado lugar na sala de aulas, mantendo as mesmas gavetas ou prateleiras durante todo o ano. Sempre que for necessário fazer alguma mudança, deve ser-lhe explicada previamente, de forma simples e cuidadosa. A maioria das crianças adaptam-se rapidamente à estrutura da sala de aulas e conseguem perceber o que se está a passar. No entanto, a criança com síndroma de Asperger pode precisar de uma ajuda adicional para conseguir acompanhar o currículo (programa).
Ambiente de linguagem e comunicação
As crianças com síndroma de Asperger costumam ter boas capacidades linguísticas, incluindo vocabulários extensos e a capacidade para utilizar estruturas gramaticais complexas. No entanto, estas capacidades são superficiais e disfarçam as dificuldades de comunicação efectivas nomeadamente na utilização social da linguagem (pragmática) e na capacidade para transmitir e compreender o significado (semântica). Estas crianças não aprendem as capacidades semânticas e pragmáticas necessárias pelo simples facto de estarem rodeadas por um ambiente de comunicação rico. O objectivo da intervenção é criar um ambiente que:
- Ajude as crianças a desenvolverem a intenção de comunicação, tanto verbal como não verbal;
- Desenvolva a capacidade da criança para iniciar e manter uma conversa;
- Aperfeiçoe a compreensão do significado por parte da criança.
A intervenção deve começar a nível da comunicação da criança – e não a nível da linguagem. Embora as suas capacidades de comunicação possam não se desenvolver pelos parâmetros habituais, a criança faz uma tentativa genuína de iniciar o processo de comunicação. As respostas que lhe damos devem reflecti-lo.
Estruturação do ambiente de linguagem
É extremamente útil simplificar e estruturar o ambiente de linguagem, tendo em conta os seguintes aspectos:
- Dirija-se à criança pelo nome antes de lhe dar uma instrução, sobretudo se estiver a dar instruções à turma enquanto grupo.
- Encoraje e reforce todas as tentativas de comunicação.
- Utilize instruções concretas, directas e explícitas – sempre que possível apoiadas por imagens.
- Se tiver de dar uma série de instruções, dê uma de cada vez.
- Dê tempo à criança para responder e, depois, verifique se ela compreendeu.
- Se necessário, repita a instrução – sem a reformular (a criança pode pensar que se trata de outra instrução).
- Ensine à criança uma expressão tipo “código” para utilizarem quando ela não compreender uma instrução – isto pode evitar alguma frustração de ambos os lados.
- Muitas vezes, a criança com síndroma de Asperger fica confusa com as perguntas. Sempre que possível, transforme-as em afirmações (p. exp.: “O tempo hoje está...” em vez de “Como é que está o tempo hoje?”).
- Reconheça as intenções da criança. Ela pode dizer “Queres batatas fritas?” quando, na realidade, é ela que quer batatas fritas.
- Oriente a criança no sentido da resposta certa para diferentes situações.
- Ao ler um texto ou ouvir outras pessoas falar, chame a atenção da criança para o modo como as palavras normalmente são encadeadas.
- Tente estar ciente da linguagem utilizada – será que a criança pode interpretá-la mal? Tenha cuidado para não utilizar sarcasmo nem ironia.
- Proponha actividades que apresentem oportunidades para as crianças se revezarem e agirem reciprocamente.
Desenvolvimento das capacidades e da compreensão da criança
- O intérprete desempenha um papel fundamental neste aspecto, na medida em que é alguém que pode ajudar a criança a compreender o mundo, mas também ajuda o mundo a compreender a criança. Esse alguém pode ser um professor, mas os colegas de turma também têm o seu papel.
- Se a criança se limitar a repetir palavras ou segmentos de linguagem (ecolalia), isso pode significar que não compreendeu alguma coisa. Também pode indiciar ansiedade. Simplifique a linguagem utilizada e procure indícios de tensão.
- Se a criança tiver áreas específicas de capacidade ou interesse, utilize-as como ponto de partida para o trabalho linguístico.
- Ajude a criança a ter consciência das necessidades do interlocutor, ensinando-a a modificar o tom de voz e o volume de acordo com a situação.
- Encoraje o contacto ocular – sem treiná-la para isso.
- A criança pode compreender os significados literais, em vez dos significados metafóricos ou implícitos. Experimente algumas metáforas comuns como, por exemplo: “Sobe as meias!” e explique-lhe o que significam. Explique bem os significados implícitos que utilizar. Pode dizer: “Está muito barulho”, mas na realidade quer dizer: “Estejam calados”.
- Ajude as crianças a compreenderem o significado e as emoções subjacentes a certas expressões faciais. Chame a sua atenção para imagens de livros e revistas que ilustrem diferentes expressões – e para o seu próprio rosto ao espelho.
O jogo de simulação pode ser útil para algumas crianças na representação de situações que envolvam reacções emocionais. O visionamento de vídeos pode ajudar a criança a reconhecer emoções através das expressões faciais, posturas e gestos. Para a maioria das crianças, a linguagem é uma maneira de entrar num mundo social estimulante. Para as crianças com síndroma de Asperger não é necessariamente assim.
Ambiente social
A dificuldade em desenvolver capacidades interpessoais fluentes é provavelmente a característica mais marcante nas crianças com síndroma de Asperger. Estas crianças não são anti-sociais. Em vez disso, são associais – às vezes querem fazer parte do mundo social, mas não sabem como entrar nele. Porém, estas crianças/jovens não adquirem capacidades sociais incidentalmente: têm de aprendê-las especificamente. A intervenção tem de começar ao nível da interacção da criança, reconhecendo que é socialmente imatura – seja qual for o seu nível de desempenho académico. É necessário não esquecer que a criança pode ser feliz nas suas actividades solitárias. Não devemos forçar a criança a participar; devemos sim adoptar a abordagem de aperfeiçoar as suas capacidades sociais. O ambiente da sala de aulas deve ter em conta a ansiedade que uma criança pode sentir por fazer parte de um grupo. Devem ser dadas oportunidades à criança para ter o seu próprio “espaço” ocasionalmente. Os outros têm de compreender as dificuldades colocadas por estas crianças/jovens e os motivos por que elas se comportam assim. Estas crianças/jovens parecem relacionar-se mais facilmente com adultos do que com outras crianças da mesma idade, possivelmente porque os adultos fazem mais concessões e modificam o próprio comportamento para com a criança.
A criança/jovem com síndroma de Asperger pode parecer ingénua e crédula, incapaz de distinguir as abordagens amigáveis das abordagens que se destinam a “dar-lhe a volta”. Os colegas parecem assumir o papel de “amigo” ou “inimigo”. A intervenção começa pela observação, identificando as crianças que agem de forma positiva e as que são potenciais focos de ansiedade. Esta ansiedade pode não ser imediatamente óbvia. A criança pode cismar com a sua percepção de um incidente – reagindo negativamente muito mais tarde. Considere os seguintes aspectos ao planear a intervenção:
- Certifique-se de que todos os funcionários estão cientes das dificuldades sociais que derivam da síndroma de Asperger e estão preparados para “fazer concessões” de uma forma consensual.
- Com o consentimento dos pais, pode ser útil falar com os outros alunos da turma/escola sobre a síndroma de Asperger.
- Como parte de uma política escolar geral em prol de uma gestão de comportamentos positiva, certifique-se de que todas as crianças estão cientes de que as brigas são inaceitáveis.
- Ensine a criança a responder a abordagens indesejadas, dado que uma criança atormentada pode tornar-se mais hostil e agressiva.
- Certifique-se de que a criança sabe a que adulto se deve dirigir quando fica transtornada.
- Pense em disponibilizar uma área tranquila para a criança se retirar quando se sentir ansiosa.
- Analise o grupo da turma e seleccione crianças mais maduras para agirem como “amigos” ou para formarem um “círculo de amigos”.
O sistema de “amigo” envolve a identificação de outra criança que esteja disposta a ajudar a criança com síndroma de Asperger a negociar as dificuldades. É possível formar “círculos de amigos” para que o ónus não recaia apenas sobre uma criança.
Desenvolvimento das capacidades e da compreensão da criança
O sentido de si próprio. Diz-se frequentemente que as crianças com síndroma de Asperger têm um elevado nível de egocentrismo. Dito desta maneira, parece que elas optam por agir assim – mas isso não é verdade! Muitas vezes, nem sequer compreendem os seus sentimentos e comportamentos. Pode acontecer a criança preocupar-se com o seu próprio bem-estar. As perguntas de adolescentes capazes como, por exemplo: “Estarei louco?” causam preocupação em todos os que os rodeiam. O objectivo da intervenção consiste em aumentar a confiança da criança em si própria enquanto indivíduo, uma vez que uma maior autoconfiança reduz a ansiedade. Devem ser tidos em conta os seguintes aspectos:
- É necessário esforçarmo-nos por fazer com que a criança tenha uma imagem positiva de si própria.
- Chegará o momento certo para comunicar à criança os antecedentes das suas dificuldades. Isto terá de ser debatido entre os pais e os profissionais.
- Depois de a criança estar ciente do “diagnóstico”, desencoraje-a de culpar a síndroma de Asperger por tudo e por nada. Encoraje-a a pensar em estratégias alternativas.
- Poderá ser necessário ensinar a criança a desenvolver um “sentido de si própria”. Encoraje-a a reflectir no seu próprio papel nos acontecimentos e nas actividades, utilizando fotografias ou vídeos e relatos subjectivos.
Interacção com terceiros. As crianças com síndroma de Asperger não estão cientes das necessidades de terceiros relativamente a elas. A intervenção visa aumentar o desejo da criança para interagir com terceiros. Pode começar por aumentar a consciência da criança relativamente ao comportamento de terceiros e, em seguida, trabalhar para ensinar a criança a interagir com terceiros. Para isso, é necessário ensinar-lhe capacidades sociais específicas, de preferência em ambientes funcionais e realistas:
- Identifique as áreas de interacção social em que a criança se debate particularmente. Analise as capacidades necessárias para melhorar o seu desempenho e ensine-lhas em pequenos passos realizáveis. Por exemplo: como entrar num grupo; como falar sobre coisas interessantes para os outros – e não só sobre os seus interesses relativamente a “animais domésticos”; como se manter envolvido no assunto do grupo.
- Tire o máximo partido de actividades que se prestem a trabalhar em equipa como, por exemplo, leitura em parceria.
- Dê oportunidade à criança para jogar jogos de tabuleiro simples como, por exemplo, damas, com um parceiro. Inicialmente, o parceiro pode ser um adulto em quem ela confie, introduzindo outra criança no jogo posteriormente.
- Comece por envolver a criança em jogos de equipa simples – na sala de aula, na aula de ginástica ou no recreio. Explique muito bem o papel da criança no jogo.
Ambiente curricular
A maioria das crianças com síndroma de Asperger frequenta estabelecimentos de ensino regular. Alguns professores sentem-se sobrecarregados com a responsabilidade de ensinar uma criança dessas numa turma de 28 alunos ou mais. Não é por nenhuma razão hostil ou negativa, mas simplesmente devido a ansiedade face ao desconhecido. Pensam que podem ter de utilizar estratégias fora do repertório habitual de ensino para satisfazer as necessidades das crianças. Na realidade, o que é necessário é uma combinação de compreensão da síndroma de Asperger e de algumas práticas recomendadas na sala de aula. Os professores têm de estar informados, ser tolerantes e estabelecer empatia numa situação em que todos os funcionários estejam cientes das implicações pedagógicas desta síndroma. As limitações específicas fazem parte de tal síndroma, mas haverá muitas capacidades que a escola pode desenvolver na criança. As dificuldades da criança com esta síndroma derivam das limitações da “Teoria da Mente”, Coerência Central e Função Executiva. Deste modo, estas crianças têm uma perspectiva diferente do mundo, o que se reflecte no seu estilo de aprendizagem. O estilo de aprendizagem destas crianças é definido pelas seguintes características:
- Motivação. A criança não tem motivos competitivos. É desprovida de orgulho e vergonha, e não tem desejo de “sobressair”.
- Imitação. Embora consiga copiar o que os outros fazem, é-lhe difícil ajustar estes movimentos copiados ao seu próprio modelo de referência.
- Percepção. Existe a possibilidade de respostas incoerentes ou inesperadas aos estímulos sensoriais.
- Atenção. O foco de atenção da criança é frequentemente reduzido e/ou obsessivo. As características dos estímulos podem ser combinadas de modo idiossincrático.
- Memória. Provavelmente, a memória da criança é episódica, isto é, os acontecimentos não são arquivados no contexto em que ocorrem. É deste modo que são arquivadas listas de factos, sem nenhuma estrutura de significado que as relacione.
- Sequenciação. A criança terá dificuldade em seguir sequências. Talvez consiga estabelecer correspondência com uma sequência e, no entanto, não consegue ir além do modelo para deduzir a regra ou o princípio em que se baseia. Por esse motivo, qualquer alteração nas sequências de acontecimentos deixa a criança angustiada por não reconhecer o princípio orientador.
- Resolução de problemas. A criança tenta aprender respostas definidas para situações concretas. Pode aprender um conjunto de estratégias, mas não está ciente desse conhecimento, pelo que não é capaz de seleccionar uma estratégia adequada para uma nova situação.
Ao percorrer o Currículo Nacional, existem temas comuns que pode ser necessário diferenciar para acomodar o estilo de aprendizagem particular da criança com síndroma de Asperger. Para o ilustrar, serão de seguida analisados, do ponto de vista de tal criança, alguns elementos seleccionados do Currículo Nacional, destacando-se exemplos de possíveis dificuldades, bem como as estratégias de intervenção sugeridas.
Área Curricular: Português
Exemplo de dificuldades
A capacidade da criança para pensar com imaginação é limitada, originando problemas de escrita criativa.
- Dê à criança oportunidades adicionais para escrever sobre experiências verídicas.
- Utilize estes relatos como base para desenvolver a escrita criativa.
- Por exemplo: “Foi isto que aconteceu realmente quando foste passear para a praia; mas o que aconteceria se tivesse chovido/tivesses perdido a semanada?”
- Ajude a criança, discutindo com ela as possibilidades. Limite os elementos criativos à experiência da criança.
Exemplo de dificuldades
- A criança pode ter inúmeras capacidades para a leitura.
- Consegue descodificar as palavras, mas não compreende exactamente aquilo que leu.
- Aumente a compreensão da criança, chamando-lhe a atenção para as ilustrações – “O que está acontecer nesta imagem?”
- Peça-lhe para prever o que vai acontecer em seguida. Por exemplo: “O que é que o menino vai fazer agora?”
- Peça-lhe para contar o que acaba de ler.
- Dê preferência a livros mais realistas do que fantasiosos.
- Dê à criança livre acesso a livros que não sejam de ficção. É-lhe mais fácil obter informação a partir destes livros do que de histórias.
Área Curricular: Educação Visual e Tecnológica (EVT)
Exemplo de dificuldades
O aluno tem dificuldade nos aspectos de EVT que exigem um nível significativo de pensamento criativo.
Escolha um projecto que seja prático, relevante para o aluno e em que ele possa utilizar uma experiência directa recente.
Exemplo de dificuldades
Para o aluno, é difícil escolher os materiais e o equipamento a utilizar.
Não sobrecarregue o aluno com demasiados itens à escolha. Não lhe apresente mais de 2 alternativas de cada vez.
Exemplo de dificuldades
O aluno distrai-se com pormenores irrelevantes. Foge completamente ao assunto e não consegue concluir a tarefa.
Minimize as distracções com a disposição dos materiais. Ajude o aluno a manter-se concentrado fornecendo-lhe uma descrição escrita, com a tarefa delineada em passos explícitos.
O currículo espera que o aluno tenha uma abordagem aberta ao desenvolver as ideias e que explore um leque de soluções possíveis antes de seleccionar uma. Este aluno talvez só veja uma possibilidade, mantendo-se estritamente fiel a ela.
Simplifique a linguagem utilizada. Dê uma instrução de cada vez. Utilize objectos e imagens para ajudar a criança a compreender.
A criança tem um fascínio por números e faz incessantemente as mesmas perguntas – interrompendo a aula.
Defina uma regra explícita. Diga à criança que só pode fazer a mesma pergunta, por exemplo, 3 vezes. Tente reservar algum tempo para que a criança possa colocar as suas dúvidas.
A criança tem dificuldade em compreender a linguagem Matemática. Fica particularmente confusa quando são utilizadas palavras (enunciados) diferentes com o mesmo significado. Por exemplo, multiplicar/vezes.
Utilize exemplos práticos para ajudar a que as palavras façam sentido. Reúna um conjunto de palavras com conceitos relacionados, que a criança possa utilizar como referência.
A criança não tem a certeza de como responder a perguntas do tipo: “Porquê?”
Sempre que possível, transforme as perguntas em afirmações, com um espaço em branco para a resposta da criança.
Área Curricular: Ciências
Exemplo de dificuldades
O aluno prefere trabalhar sozinho e resiste a ter de partilhar uma tarefa com outro aluno. As actividades de grupo apresentam ainda mais dificuldades.
Estratégias sugeridas
Seleccione cuidadosamente os parceiros e os membros dos grupos. Explique bem o papel de cada parceiro/membro do grupo. Inicialmente, talvez o aluno precise de ter um papel passivo como, por exemplo, registar os resultados. Gradualmente, prepare-o para ter um papel mais activo.
O aluno não consegue mostrar consideração pela sua própria segurança nem pela dos outros.
O aluno não consegue mostrar consideração pela sua própria segurança nem pela dos outros.
É importante descrever da forma mais explícita as possíveis consequências das diferentes acções. É preciso ensiná-los a verificarem os procedimentos e incutir-lhes essa rotina.
O aluno é incapaz de pedir ajuda nas aulas.
Primeiro, ensine-o a reconhecer que está “encravado”. Uma vez que ele não consegue “pôr-se no lugar” do professor, talvez não se dê conta de que este dispõe das informações de que ele precisa. Ensine-lhe especificamente como pedir a ajuda de que necessita.
Para muitas crianças com síndroma de Asperger, não é o currículo que apresenta mais dificuldades, mas sim as áreas não curriculares (p. exp.: o recreio, o intervalo para o almoço), que lhes colocam mais desafios e com que têm maior dificuldade em lidar. É o seu comportamento nestas alturas que pode começar por suscitar preocupações nos professores que podem julgar que o seu papel principal é ensinar o programa (currículo) e sentir-se menos seguros na sua capacidade para desenvolver as capacidades interpessoais da criança na medida necessária para uma criança com tal síndroma. As seguintes informações visam apresentar pontos de partida de intervenção, proporcionando exemplos de práticas recomendadas numa série de situações diferentes.
Recreio
Para a maioria das crianças, o recreio é a melhor parte do dia na escola, mas as crianças/jovens com síndroma de Asperger podem recear a hora do recreio. O recreio não é estruturado. As crianças são ruidosas e exuberantes. Não há regras. Formam-se pares e grupos. Os professores relatam frequentemente que a criança com esta síndroma não tem amigos e se comporta como um solitário no recreio. O recreio apresenta oportunidades para desenvolver as capacidades sociais da criança – desde que aceite a necessidade da criança para relaxar à sua maneira. As estratégias para ajudar a criança/jovem com este síndroma a lidar com o recreio incluem:
- Definir um “espírito” de recreio, em que se encoraja a colaboração.
- Aceitar que a criança com tal síndroma pode precisar de um tempo só para si durante o intervalo – como se fosse uma pausa das exigências sociais da sala de aula.
- Organizar jogos sociais simples e estruturados – em que o papel de cada indivíduo é óbvio. - Convidar a criança a participar.
- Encorajar a criança a observar as actividades que decorrem no recreio. Explicar essas tarefas enquanto decorrem. Arranjar uma actividade que seja apelativa para a criança e, com o apoio dos colegas, encorajá-la a participar.
- Ensinar à criança “frases introdutórias” que a ajudem a iniciar uma conversa. Muitas vezes, estas crianças/jovens até querem participar nos acontecimentos – só não sabem como fazê-lo.
Movimentar-se pela escola
No 1.º ciclo há muito movimento e espaço para muita confusão em determinadas alturas do dia. Este movimento e esta confusão aumentam significativamente nos 2.º e 3.º Ciclos e Secundário, em que existe frequentemente um movimento colectivo (do tipo “manada”) no fim de cada aula. Estas alturas podem ser de grande tensão para a criança com síndroma de Asperger, simplesmente devido ao elevado número de crianças que se deslocam numa pequena área. Além disso, os 2.º e 3.º Ciclos e Secundário acarretam outra fonte de ansiedade com a necessidade de encontrar a sala certa num edifício enorme. As estratégias para ajudar a criança com esta síndroma a lidar com estas movimentações pela escola incluem:
- Inicialmente, desfasar a chegada/partida da criança para que o volume de “tráfego” seja menos arrasador.
- Preparar as visitas às escolas dos 2.º e 3.º Ciclos e Secundário antes da transferência, para dar uma oportunidade à criança para aprender a respectiva planta.
- Estabelecer uma rede de “amigos” ou um “círculo de amigos” que estejam disponíveis para servir de “guia”.
Na cantina
As cantinas podem ser espaços muito barulhentos e muito exigentes do ponto de vista social. Espera-se que as crianças comam em público ao lado de outras seis ou oito crianças. Na maioria das escolas, a hora do almoço envolve estar na fila – uma actividade que muitas crianças com síndroma de Asperger têm dificuldade em tolerar. As estratégias para ajudar a criança/jovem a lidar com a cantina da escola incluem:
- Estabelecer regras claras, reforçadas por auxiliares visuais.
- Simular as rotinas do almoço no refeitório vazio e sossegado.
- Considerar permitir que a criança passe para o princípio ou fim da fila, em vez de ter de ficar no meio de toda a gente. Deste modo, a criança sente-se menos ameaçada.
- Alertar os auxiliares do refeitório para as dificuldades da criança e as estratégias que estão a ser utilizadas.
- Ensinar à criança capacidades de conversação simples para o ajudar a integrar-se com as outras crianças à mesa.
Pedir ajuda e resolver problemas
“Stôra! Como é que eu faço isto?” A maioria das crianças pede ajuda prontamente quando não tem a certeza de alguma coisa. As crianças com síndroma de Asperger têm muita dificuldade em fazê-lo – por uma série de motivos:
- As crianças/jovens com tal síndroma têm dificuldade em resolver problemas – em combinar os elementos de uma tarefa para as ajudarem a chegar a uma conclusão. Pedir ajuda é um dos elementos do processo de resolução de problemas. Não se apercebem necessariamente da relação entre o problema e a ajuda externa.
- Devido à capacidade limitada que estas crianças/jovens têm para reconhecer a existência de diferentes pontos de vista, podem não se aperceber de que outra pessoa possa ter uma solução para o problema com que se deparam.
Infelizmente, este comportamento pode ser confundido com preguiça ou falta de motivação – se pensarmos que a criança/jovem não quer trabalhar, e não que simplesmente não consegue avançar. As estratégias para ajudar estas crianças/jovens a pedir ajuda incluem:
- Ter consciência das tarefas em que a criança tem mais dificuldades.
- Se a criança tiver dificuldade em interpretar uma tarefa, experimente trabalhar com ela (sentado ao lado dela). Execute primeiro a tarefa e chame a atenção da criança para aquilo que está a fazer. Deixe-a experimentar fazer uma parte da tarefa. Reduza gradualmente a sua participação até a criança já conseguir trabalhar sozinha.
- Quando a criança tiver concluído a tarefa, faça-a reflectir naquilo que aprendeu, explicando-lhe aquilo que fez e como o fez.
- Ensine-a especificamente a reconhecer quando está “encravada” e como pode pedir ajuda.
Trabalhar com outros
Os professores esperam que as crianças trabalhem em grupo com outras crianças várias vezes durante o dia. Podem não se dar conta da tensão que isto pode causar numa criança com síndroma de Asperger, que tem tanta dificuldade em relacionar-se com os outros. Esta criança/jovem tem pouca ou nenhuma consciência dos sentimentos dos outros ou do impacto do seu próprio comportamento nos outros. Pode aceitar passivamente a presença de outras crianças, mas algumas crianças ficam tensas simplesmente porque estão sentadas muito perto de alguém. O contacto é unilateral – da outra criança para com ela, mas raramente é recíproco. Além disso, na escola espera-se que as crianças trabalhem em grupo (com um parceiro ou com um pequeno grupo). Com algum planeamento e preparação cuidadosos, a criança com síndroma de Asperger pode ser incluída nestas actividades. As estratégias para ajudar estas crianças/jovens a trabalhar com outros incluem:
- Ter consciência do nível de contacto social que a criança consegue tolerar sem ficar ansiosa.
- Inicialmente, permitir que a criança se sente “na ponta” durante uma actividade de grupo, possivelmente com um professor de educação especial entre ela e a criança do lado.
- Considerar dispor os lugares de uma determinada maneira. A criança pode sentir-se mais à vontade se a criança “do lado” se sentar na diagonal à frente dela – e não ao lado.
- Quando a criança já tolerar que outras crianças se sentem ao seu lado, o professor pode preparar tarefas simples em que tenham de revezar-se – inicialmente só com ela; mais tarde, com outra criança; por último, o professor retira-se, encorajando a criança a trabalhar com o parceiro.
- É possível que surjam oportunidades para utilizar as próprias capacidades ou interesses especiais da criança, talvez conseguindo que ela mostre a um parceiro como fazer alguma coisa no computador.
- Definir explicitamente e ensinar o papel de cada um numa tarefa de grupo ou numa actividade colectiva – acabando com a incerteza para a criança com tal síndroma.
A vida escolar é mais simples para as crianças com síndroma de Asperger quando os adultos que as rodeiam reconhecem até que ponto as exigências sociais resultam em tensão. A intervenção e o apoio nesta área dão frutos em termos de percepção das regras sociais aplicáveis ao ensino, dado que a tensão obsta à aprendizagem.
Em resumo:
A síndroma de Asperger é uma condição que se pensa estar enquadrada no espectro do autismo – com traços distintivos suficientes para garantir um “rótulo” próprio.
- Foi descrita pela primeira vez em 1944 pelo médico austríaco Hans Asperger, cujo trabalho foi publicado pela primeira vez em inglês em 1991.
- Caracteriza-se por limitações subtis nas três áreas de desenvolvimento: comunicação social, interacção social e imaginação social. Em certos casos, também se registam problemas adicionais de organização e coordenação motora.
- Afecta pessoas de inteligência média e acima da média.
- Pensa-se que a prevalência se situe na ordem de 36 por 10.000.
- A probabilidade de incidência é maior nos rapazes do que nas raparigas, com uma taxa de probabilidade de 10 rapazes para 1 rapariga.
- Hans Asperger considerou que a “formação” pedagógica ajudaria a criança/jovem e, para a maioria das crianças/jovens, provou-se ser verdade. Para ser eficaz, a intervenção para as crianças/jovens com síndroma de Asperger tem de assentar na compreensão da natureza de condição e das limitações fundamentais inerentes. É necessário fazer concessões em função da individualidade, uma vez que cada criança/jovem é afectada de maneira diferente.
Finalmente:
- Comece no nível da criança/jovem.
- Tente ver o mundo do ponto de vista da criança/jovem.
- Adapte o ambiente escolar de modo a facilitar a aprendizagem por parte da criança/jovem.
- Consulte os Pais e o professor de educação especial e colabore com eles.
- Considere intervenções específicas para desenvolver as capacidades da criança/jovem nas áreas de Interacção Social, Comunicação Social e Pensamento Flexível.
- Familiarize-se com as explicações psicológicas subjacentes ao estilo de aprendizagem da criança/jovem.

Bibliografia:
Attwood, T. (2006). O Síndrome de Asperger: Um guia para pais e profissionais. Lisboa: Verbo Editora.
Tucker, E. (S/d). Síndrome Asperger: Guia para professores. Associação Portuguesa do Síndrome de Asperger.
Disponível em: http://www.apsa.org.pt/
Cumine, V.; Leach, J.; Stevenson, G. (2006). Compreender a Síndroma de Asperger: Guia prático para educadores. Porto: Porto Editora
Kirby, B.L. (S/d). Asperger's Syndrome Guide For Teachers. Disponível em: http://www.udel.edu/bkirby/asperger/
A Síndrome
de Asperger é uma perturbação neurocomportamental de base genética,
pode ser definida como uma perturbação do desenvolvimento que se
manifesta por alterações sobretudo na interacção social na comunicação e
no comportamento. Embora seja uma disfunção com origem num
funcionamento cerebral particular, não existe marcador biológico, pelo
que o diagnóstico se baseia num conjunto de critérios comportamentais.
Entre as características mais comuns podemos destacar:
Entre as características mais comuns podemos destacar:
- Défice de comportamento social;
- Interesses limitados;
- Comportamentos rotineiros;
- Peculiaridade do discurso e da linguagem;
- Perturbação na comunicação não verbal;
- Descoordenação motora.
Como consequência destas dificuldades os portadores de Síndrome de Asperger acabam por se isolar e limitar os seus interesses a determinados temas assuntos, atitude que prejudica ainda mais a sua relação com o outro. Calcula-se que em Portugal existam cerca de 40.000 portadores de Síndrome de Asperger afectando maioritariamente os rapazes.
O Diagnóstico precoce é essencial para proporcionar aos portadores, os recursos necessários e a que têm direito que lhes permitam atingir o seu potencial, o qual muitas vezes é extraordinário, como pessoas verdadeiramente integradas na sociedade.
in http://www.apsa.org.pt/
CARACTERÍSTICAS PRINCIPAIS
Origem
A primeira pesquisa neste campo foi realizada pelo psicólogo austríaco, Hans Asperger, em 1943 – mas apenas nos últimos 20 anos aumentou a informação disponível e o conhecimento da classe médica e do público em geral sobre o que se entendeu chamar Síndroma de Asperger. Para isto muito contribui o trabalho de especialistas como Tony Attwood, Lorna Wing, entre outros. O Síndroma de Asperger foi oficialmente classificado nos EUA como uma disfunção psicológica em 1994 que afecta o modo como uma pessoa comunica e se relaciona com os outros. Entre outras características das crianças/jovens com este Síndroma, podemos destacar as seguintes:
- Dificuldade na comunicação;
- Dificuldade no relacionamento social;
- Dificuldade no pensamento abstracto.
As pessoas com síndrome de Asperger têm problemas de linguagem em menor escala do que as classificadas como autistas, falam mais fluentemente e não têm dificuldades de aprendizagem tão marcadas. Têm normalmente inteligência (Q.I.) média ou mesmo acima da média e muitas não são diagnosticadas como tal, sendo muitas vezes referidas, pela família e professores, como estranhos, excêntricos, originais, diferentes, extravagantes ou esquisitos. Os casos menos pronunciados, podem fazer excelentes progressos, ter sucesso, e mesmo continuar os estudos ao nível universitário e arranjar um emprego.
Interacção Social
Ao contrário dos autistas “clássicos”, que normalmente estão ausentes e desinteressados do mundo que os rodeia, muitas crianças com síndroma de Asperger querem ser sociáveis e gostam do contacto humano. Têm no entanto dificuldade em perceber sinais não-verbais, incluindo os sentimentos traduzidos em expressões faciais, o que levanta problemas em criar e manter relações com pessoas que não percebem esta dificuldade. Precisam de aprender aspectos do convívio social que nós adquirimos sem pensar, como a entoação da voz, linguagem corporal e expressões faciais. Essencialmente, a criança com síndroma de Asperger:
- Isola-se socialmente, mas pode não se preocupar com isso;
- Pode ficar tensa e agitada ao tentar lidar com as abordagens e as exigências sociais de terceiros;
- Começa a ter consciência de que os seus colegas têm amizades, sobretudo quando atinge a adolescência. Nessa altura, pode querer ter os seus amigos, mas não tem nenhuma estratégia para desenvolver e consolidar amizades;
- Tem dificuldade em seguir as “deixas” sociais;
- Pode ter um comportamento socialmente inaceitável.
Comunicação em contextos sociais
- Pode ter uma linguagem aparentemente perfeita, mas com tendência para ser formal e pedante. “Como está? Chamo-me João” pode ser uma saudação típica de um adolescente com síndroma de Asperger – mas é precisamente isso que o distancia dos seus colegas, expondo-o ao ridículo.
- Tem frequentemente uma voz sem expressão (monocórdica). Também pode ter dificuldade em interpretar as diferentes entoações de terceiros. Quase toda a gente sabe dizer se uma pessoa está zangada, aborrecida ou radiante apenas pela entoação (ou inflexão de voz). Muitas vezes, a criança com síndroma de Asperger não consegue ter este tipo de discernimento, o que pode originar algumas situações complicadas. Por exemplo, um professor teve de combinar um sinal visual com um aluno – “Quando eu tirar os óculos enquanto estiver a olhar para ti, já sabes que estou zangado contigo”. Levantar a voz não tinha nenhum efeito sobre a criança.
- Também pode ter dificuldade em utilizar e interpretar comunicação não verbal como, por exemplo, linguagem corporal, gestos e expressões faciais.
- Pode compreender os outros de forma muito literal. Um exemplo: após o Rui se ter recusado terminantemente a ajudar a avó a arrumar a cozinha, esta disse-lhe: “Deves pensar que tens o rei na barriga!” O Rui respondeu-lhe: “Na barriga? Não é possível engolir um rei!”, não percebendo que a avó se referia à sua teimosia em não a ajudar. A criança não reagiria a uma expressão como “Está calor aqui” – enquanto todas as outras pessoas perceberiam de imediato o sinal para abrir uma janela.
Pensamento abstracto
Podem ser excelentes na memorização de factos e números mas têm normalmente dificuldade ao nível do pensamento abstracto. Isto é causa frequente de problemas na aprendizagem, em ambiente escolar, de matérias como o português ou filosofia. No entanto, podem ser excelentes a matemática ou geografia.
Interesses especiais
As crianças com síndroma de Asperger desenvolvem interesses obsessivos e podem em consequência disto adquirir um conhecimento enciclopédico sobre determinada matéria. Podem ficar fascinados com horários de comboios, um certo programa de televisão ou previsões meteorológicas. Isto pode originar-lhes alguma frustração por não entenderem que os seus interesses não são partilhados pelos outros. No entanto, estas obsessões podem ser aproveitadas, e conduzir a boas oportunidades profissionais e de investigação.
Gosto por Rotinas
Não gostam de alterações ou mudanças. Podem impor as suas rotinas, tais como insistir em seguir sempre o mesmo caminho para a escola. Na escola podem ficar nervosos com uma alteração no horário, ou mudança de professor. Gostam normalmente de ter uma rotina diária coerente e imutável. Se trabalham de acordo com um horário, um atraso inesperado, devido a um demora nos transportes ou a problemas de tráfego, podem torná-los muito nervosos ou ansiosos.
Sensibilidade Sensorial
Têm uma resposta exagerada a alguns estímulos, podendo ser afectados quaisquer dos 5 sentidos. Por exemplo, podem ter vómitos com um determinado sabor ou cheiro, podem não suportar que lhe mexam na cabeça, podem não gostar de tocar em determinadas texturas, podem não suportar determinados sons por demasiado intensos ou agudos.
Coordenação Motora
São normalmente desajeitados e têm dificuldade na coordenação motora, (atar os sapatos, andar de bicicleta, etc.). A sua forma de andar é peculiar. Essencialmente, a criança/jovem com tal síndroma:
- Pode ter movimentos bruscos e desastrados;
- Tem frequentemente problemas de organização – não consegue orientar-se nem reunir o material de que precisa;
- Tem dificuldade em escrever e desenhar ordenadamente e, muitas vezes, não termina as tarefas.
Vulnerabilidades
As crianças e adultos com síndroma de Asperger são muito vulneráveis. A adolescência amplifica a sua luta interna, gerando grande ansiedade. A possibilidade de suicídio deve ser uma preocupação. Problemas que podem surgir englobam, entre outros: vulnerabilidade ao abuso, (praxe, gozo, exploração, etc), porque os seus comportamentos são vistos como excêntricos ou peculiares, têm dificuldade de adaptação e encontram-se sozinhos; deficiente visão de conjunto devido ao seu interesse no detalhe (em consequência, não conseguem antever os resultados das suas acções ou palavras, nem colocar os assuntos em contexto); grande ansiedade quando há alterações da rotina do dia-a-dia, (p. exp., atraso de um autocarro, mudança de professor, desaparecimento da marca favorita, etc.); dificuldade em planear porque isso requer a capacidade de pensar em hipóteses e prever consequências; dificuldade em expressar os seus pensamentos, em comunicar os seus medos, os seus problemas e frustrações.
O que causa a Síndroma de Asperger?
As
causas do autismo e da síndrome de Asperger não são ainda totalmente
compreendidas. Muitos especialistas acreditam que as alterações do
comportamento que constituem a síndroma de Asperger podem não resultar
de uma única causa. Existe alguma informação que leva a pensar ser a
síndroma de Asperger é provocado por um conjunto de factores
neuro-biológicos que afectam o desenvolvimento cerebral, e não ser
devida, como se chegou a pensar, a privação de afecto, ou à criança ter
crescido num ambiente demasiado austero. Hoje em dia, a síndroma de
Asperger é descrita como uma disfunção cerebral e os investigadores
procuram apontar uma ou mais áreas do cérebro em que esta disfunção
ocorre. Com os avanços da tecnologia, talvez seja possível concretezar
melhor esta ideia.
Perspectivas de Futuro?
Actualmente não existem em Portugal instituições dedicadas exclusivamente às crianças com síndroma de Asperger. Algumas andam nas escolas do ensino regular, onde o seu progresso depende do ambiente gerado à sua volta e do apoio e encorajamento de pais e professores. Outras frequentam Unidades de Intervenção Especializada, vocacionadas para crianças com problemas mais graves de desenvolvimento. As crianças/jovens com esta síndroma são mais vulneráveis porque, por um lado, podem não ter sido devidamente diagnosticadas, por outro, porque os seus problemas de aprendizagem são menos óbvios do que os de outras crianças. São por isso, normalmente, um alvo preferencial do abuso físico e verbal por parte dos seus colegas, o que os pode tornar especialmente frustrados ou angustiados. Ao crescerem tomam melhor consciência da sua diferença e podem ter tendência para a solidão e depressão. Normalmente querem ser sociáveis mas têm dificuldade em criar e manter amizades, mas o futuro não necessita de ser obrigatoriamente negro. Em adultos podem ter grande sucesso nas carreiras que escolhem, potenciando as suas qualidades de obstinação, memória e facilidade para a matemática, e podem desenvolver amizades duradouras. Como trabalhadores estes indivíduos têm características muito prezadas – pontualidade, fiabilidade e dedicação – no entanto é essencial que o ambiente de trabalho que o rodeia seja harmonioso e compreenda às suas características.
INTERVENÇÃO PEDAGÓGICA
O papel do(s) professor(s) da turma é central para a educação da criança/jovem com síndroma de Asperger. Cabe-lhe(s) assegurar que todos os alunos da turma são educados a um nível que seja adequado às necessidades de cada um. Para isso, é necessário criar um ambiente que encoraje a valorização dos indivíduos e reconheça os diferentes estilos de aprendizagem inerentes a cada um. Esse ambiente baseia-se na compreensão das necessidades de todos os alunos incluindo as crianças/jovens com síndroma de Asperger. As áreas específicas sobre as quais o professor da turma se deve debruçar são:
- Criação de um ambiente de trabalho calmo;
- Garantia de que a estrutura da sala de aulas está perfeitamente definida;
- Modificação das tarefas para tirar partido e consolidar as forças da criança;
- Garantia de que a criança compreende o que se espera dela;
- Introdução gradual da escolha, encorajando a tomada de decisões;
- Estratificação das tarefas, aumentando gradualmente as exigências feitas à criança;
- Orientação da atenção da criança a nível individual, em vez de se basear em instruções dadas a toda a turma;
- Acesso à formação disponível; Planeamento de Adequações Curriculares;
- Registo e monitorização dos progressos;
- Avaliação de estratégias de intervenção;
- Trabalho em estreita relação com a rede de apoio disponível;
- Estabelecimento e manutenção de ligações casa/escola.
Embora possa parecer inicialmente um conjunto de expectativas estimulante, na realidade não passa de um simples exemplo de práticas recomendadas na sala de aula. Um pensamento importante a não esquecer é que a criança com síndroma de Asperger faz parte do todo da comunidade escolar e deve ser aceite e apoiada por toda a comunidade escolar. Muitas vezes, estas crianças sentem-se mais à vontade a desempenhar tarefas de aprendizagem do que a brincar. Pode ser fácil lidar com a rotina e a estrutura explícita de uma tarefa de aprendizagem, desde que não seja essencial a interacção social com outras crianças. Devido à natureza subtil e penetrante da limitação, é na banalidade da existência momento a momento que podem ocorrer dificuldades para a criança com síndroma de Asperger. Deste modo, o professor tem de estar preparado para actuar a um nível igualmente subtil e penetrante, para estar presente na construção do dia da criança, para fazer parte do ambiente “protésico”, para fazer de tradutor, intermediário e, sempre que possível:
- Compreender a capacidade limitada da criança para interpretar as “deixas” sociais;
- Interpretar situações para a criança;
- Mostrar à criança o que se espera dela;
- Ajudar e ensinar as capacidades adequadas de interacção social como, por exemplo, revezar-se;
- Orientar os colegas no modo de interagir com o aluno, solicitando a sua ajuda;
- Compreender as dificuldades subtis da linguagem e da comunicação;
- Prestar atenção (“ouvir”) o padrão de utilização da linguagem da criança e estar alerta para as dificuldades de interpretação;
- Explicar, mostrar e esclarecer a criança em caso de confusão;
- Ajudar a criança a desenvolver urna utilização adequada da linguagem e a desenvolver uma consciência;
- Compreender as origens da rigidez e obsessão no comportamento da criança;
- Antecipar o que irá causar ansiedade e fazer as alterações necessárias;
- Analisar e simplificar situações ou actividades que causem alarme;
- Tornar os procedimentos associados a tarefas visualmente explícitos, através de estímulos visuais e pictóricos;
- Prestar apoio à criança nas actividades físicas, caso haja problemas de destreza;
- Ajudar a simplificar as tarefas de redacção;
- Estar preparado para os períodos de ansiedade com actividades adequadas de redução de tensão (stress);
- Avaliar regularmente o potencial para aumentar a autonomia da criança;
- Identificar as falhas no desenvolvimento de capacidades de autoajuda em áreas como vestir-se e lavar-se. Integrá-las no programa da criança;
- Identificar qualquer dificuldade de organização e produzir auxiliares práticos e visuais para ajudar a criança;
- Prestar apoio na ligação casa/escola e registar os progressos;
- Propor estratégias adequadas de recompensa nas Adequações Curriculares;
- Saber quando e como pedir ajuda ao docente de educação especial;
- Agir como elo de ligação regular com o docente de educação especial;
- Desenvolver e manter sistemas úteis de monitorização, avaliação e registo.
Além disso, o professor tem de ser:
- Calmo;
- Positivo;
- Coerente.
Principais dicas para os professores utilizarem com as crianças com síndroma de Asperger:
Comunicação
- Simplifique a linguagem utilizada.
- Dê uma instrução de cada vez, em vez de uma série de instruções.
- Mantenha as expressões faciais e os gestos simples e explícitos.
- Dê tempo à criança para responder. Utilize auxiliares visuais adicionais para ajudar a criança a compreender.
- Seja sensível às tentativas da criança para comunicar.
- Prepare situações que encorajem a criança a tentar comunicar.
Interacção social
- Compreenda que a criança pode sentir-se ameaçada pela proximidade extrema de terceiros – sobretudo de outras crianças da mesma idade.
- Permita que a criança se isole.
- Acompanhe o ritmo da criança ao tentar desenvolver uma interacção – talvez seja necessário “regredir” em termos de desenvolvimento.
- Identifique as preferências e as antipatias das crianças a nível social – utilize este conhecimento ao planear as actividades.
- É mais provável que a criança interaja com pessoas conhecidas, por isso dê-lhe tempo para ficar a conhecê-lo e não a confunda com muitas mudanças de pessoal.
Comportamento
- Adopte uma abordagem com a máxima coerência.
- Ajude a criança a compreender o que se espera dela através de rotinas explícitas e previsíveis.
- Introduza a mais pequena alteração de forma gradual.
- Ajude a explicar as mudanças através de auxiliares visuais.
- Se a criança ficar agitada, compreenda que as estratégias habituais para acalmar uma criança (por exemplo, tentar sentá-la junto a si) podem ter o efeito oposto e ela acabar por ficar ainda mais agitada.
- Se a criança tiver uma obsessão, não tente detê-la. Com o tempo, talvez consiga limitá-la – entretanto, utilize-a de forma positiva.
Em geral
- Os resultados e os progressos podem ser lentos – não desista! (Muitas vezes, construir uma relação demora muito tempo.)
- Cada criança é única – o que resulta para uma pode não resultar para outra.
- Cada criança é instável – por isso, se ela estiver num “dia não”, não pense que a culpa é sua.
- Se tudo o resto falhar, deixe-a sozinha. Amanhã é outro dia!
As palavras-chave da intervenção são rotina, clareza e coerência. As pequenas mudanças no ambiente da sala de aulas, que simplifiquem a organização e a estrutura da sala e das tarefas, podem ajudar a criança a perceber quais são as expectativas. É possível fazer alterações úteis nas seguintes áreas do ambiente:
- Ambiente físico e sensorial;
- Ambiente de linguagem e comunicação;
- Ambiente social;
- Ambiente curricular.
Em seguida, é possível integrar intervenções específicas que permitam à criança desenvolver capacidades e compreensão nas áreas de Interacção Social, Comunicação Social e Imaginação e Jogo Social. Por outras palavras, é necessário que a abordagem seja compensatória, mas também terapêutica.
Ambiente físico e sensorial
Um ambiente congestionado, imprevisível e em constante mudança só vai confundir a criança com síndroma de Asperger e deixá-la ansiosa. Através da análise do ambiente de aprendizagem da criança, é possível imaginar as modificações que poderão ajudá-la.
Organização e estrutura
A maioria das salas de aula é organizada do ponto de vista social. O papel da compreensão social é assumido pelos professores que podem, efectivamente, não ter consciência desse papel na organização da sala de aula, por exemplo, no trabalho em grupo, na partilha de material, em ouvir uma instrução enquanto grupo. Imensas salas de aula da J. Infância e 1.º ciclo são organizadas por grupos, sendo que os grupos se movem pela sala de aulas para aceder às tarefas. Poderá ser necessário permitir que a criança tenha o seu próprio “espaço” algumas vezes durante o dia. Isto é particularmente importante sempre que é introduzida uma nova tarefa, uma vez que a tensão de fazer parte de um grupo pode limitar a compreensão. Este “espaço” não tem de ser um posto de trabalho individual sofisticado – uma carteira junto à mesa do grupo, de costas para as distracções, pode ser extremamente eficaz. É útil se a criança ocupar habitualmente um determinado lugar na sala de aulas, mantendo as mesmas gavetas ou prateleiras durante todo o ano. Sempre que for necessário fazer alguma mudança, deve ser-lhe explicada previamente, de forma simples e cuidadosa. A maioria das crianças adaptam-se rapidamente à estrutura da sala de aulas e conseguem perceber o que se está a passar. No entanto, a criança com síndroma de Asperger pode precisar de uma ajuda adicional para conseguir acompanhar o currículo (programa).
Ambiente de linguagem e comunicação
As crianças com síndroma de Asperger costumam ter boas capacidades linguísticas, incluindo vocabulários extensos e a capacidade para utilizar estruturas gramaticais complexas. No entanto, estas capacidades são superficiais e disfarçam as dificuldades de comunicação efectivas nomeadamente na utilização social da linguagem (pragmática) e na capacidade para transmitir e compreender o significado (semântica). Estas crianças não aprendem as capacidades semânticas e pragmáticas necessárias pelo simples facto de estarem rodeadas por um ambiente de comunicação rico. O objectivo da intervenção é criar um ambiente que:
- Ajude as crianças a desenvolverem a intenção de comunicação, tanto verbal como não verbal;
- Desenvolva a capacidade da criança para iniciar e manter uma conversa;
- Aperfeiçoe a compreensão do significado por parte da criança.
A intervenção deve começar a nível da comunicação da criança – e não a nível da linguagem. Embora as suas capacidades de comunicação possam não se desenvolver pelos parâmetros habituais, a criança faz uma tentativa genuína de iniciar o processo de comunicação. As respostas que lhe damos devem reflecti-lo.
Estruturação do ambiente de linguagem
É extremamente útil simplificar e estruturar o ambiente de linguagem, tendo em conta os seguintes aspectos:
- Dirija-se à criança pelo nome antes de lhe dar uma instrução, sobretudo se estiver a dar instruções à turma enquanto grupo.
- Encoraje e reforce todas as tentativas de comunicação.
- Utilize instruções concretas, directas e explícitas – sempre que possível apoiadas por imagens.
- Se tiver de dar uma série de instruções, dê uma de cada vez.
- Dê tempo à criança para responder e, depois, verifique se ela compreendeu.
- Se necessário, repita a instrução – sem a reformular (a criança pode pensar que se trata de outra instrução).
- Ensine à criança uma expressão tipo “código” para utilizarem quando ela não compreender uma instrução – isto pode evitar alguma frustração de ambos os lados.
- Muitas vezes, a criança com síndroma de Asperger fica confusa com as perguntas. Sempre que possível, transforme-as em afirmações (p. exp.: “O tempo hoje está...” em vez de “Como é que está o tempo hoje?”).
- Reconheça as intenções da criança. Ela pode dizer “Queres batatas fritas?” quando, na realidade, é ela que quer batatas fritas.
- Oriente a criança no sentido da resposta certa para diferentes situações.
- Ao ler um texto ou ouvir outras pessoas falar, chame a atenção da criança para o modo como as palavras normalmente são encadeadas.
- Tente estar ciente da linguagem utilizada – será que a criança pode interpretá-la mal? Tenha cuidado para não utilizar sarcasmo nem ironia.
- Proponha actividades que apresentem oportunidades para as crianças se revezarem e agirem reciprocamente.
Desenvolvimento das capacidades e da compreensão da criança
- O intérprete desempenha um papel fundamental neste aspecto, na medida em que é alguém que pode ajudar a criança a compreender o mundo, mas também ajuda o mundo a compreender a criança. Esse alguém pode ser um professor, mas os colegas de turma também têm o seu papel.
- Se a criança se limitar a repetir palavras ou segmentos de linguagem (ecolalia), isso pode significar que não compreendeu alguma coisa. Também pode indiciar ansiedade. Simplifique a linguagem utilizada e procure indícios de tensão.
- Se a criança tiver áreas específicas de capacidade ou interesse, utilize-as como ponto de partida para o trabalho linguístico.
- Ajude a criança a ter consciência das necessidades do interlocutor, ensinando-a a modificar o tom de voz e o volume de acordo com a situação.
- Encoraje o contacto ocular – sem treiná-la para isso.
- A criança pode compreender os significados literais, em vez dos significados metafóricos ou implícitos. Experimente algumas metáforas comuns como, por exemplo: “Sobe as meias!” e explique-lhe o que significam. Explique bem os significados implícitos que utilizar. Pode dizer: “Está muito barulho”, mas na realidade quer dizer: “Estejam calados”.
- Ajude as crianças a compreenderem o significado e as emoções subjacentes a certas expressões faciais. Chame a sua atenção para imagens de livros e revistas que ilustrem diferentes expressões – e para o seu próprio rosto ao espelho.
O jogo de simulação pode ser útil para algumas crianças na representação de situações que envolvam reacções emocionais. O visionamento de vídeos pode ajudar a criança a reconhecer emoções através das expressões faciais, posturas e gestos. Para a maioria das crianças, a linguagem é uma maneira de entrar num mundo social estimulante. Para as crianças com síndroma de Asperger não é necessariamente assim.
Ambiente social
A dificuldade em desenvolver capacidades interpessoais fluentes é provavelmente a característica mais marcante nas crianças com síndroma de Asperger. Estas crianças não são anti-sociais. Em vez disso, são associais – às vezes querem fazer parte do mundo social, mas não sabem como entrar nele. Porém, estas crianças/jovens não adquirem capacidades sociais incidentalmente: têm de aprendê-las especificamente. A intervenção tem de começar ao nível da interacção da criança, reconhecendo que é socialmente imatura – seja qual for o seu nível de desempenho académico. É necessário não esquecer que a criança pode ser feliz nas suas actividades solitárias. Não devemos forçar a criança a participar; devemos sim adoptar a abordagem de aperfeiçoar as suas capacidades sociais. O ambiente da sala de aulas deve ter em conta a ansiedade que uma criança pode sentir por fazer parte de um grupo. Devem ser dadas oportunidades à criança para ter o seu próprio “espaço” ocasionalmente. Os outros têm de compreender as dificuldades colocadas por estas crianças/jovens e os motivos por que elas se comportam assim. Estas crianças/jovens parecem relacionar-se mais facilmente com adultos do que com outras crianças da mesma idade, possivelmente porque os adultos fazem mais concessões e modificam o próprio comportamento para com a criança.
A criança/jovem com síndroma de Asperger pode parecer ingénua e crédula, incapaz de distinguir as abordagens amigáveis das abordagens que se destinam a “dar-lhe a volta”. Os colegas parecem assumir o papel de “amigo” ou “inimigo”. A intervenção começa pela observação, identificando as crianças que agem de forma positiva e as que são potenciais focos de ansiedade. Esta ansiedade pode não ser imediatamente óbvia. A criança pode cismar com a sua percepção de um incidente – reagindo negativamente muito mais tarde. Considere os seguintes aspectos ao planear a intervenção:
- Certifique-se de que todos os funcionários estão cientes das dificuldades sociais que derivam da síndroma de Asperger e estão preparados para “fazer concessões” de uma forma consensual.
- Com o consentimento dos pais, pode ser útil falar com os outros alunos da turma/escola sobre a síndroma de Asperger.
- Como parte de uma política escolar geral em prol de uma gestão de comportamentos positiva, certifique-se de que todas as crianças estão cientes de que as brigas são inaceitáveis.
- Ensine a criança a responder a abordagens indesejadas, dado que uma criança atormentada pode tornar-se mais hostil e agressiva.
- Certifique-se de que a criança sabe a que adulto se deve dirigir quando fica transtornada.
- Pense em disponibilizar uma área tranquila para a criança se retirar quando se sentir ansiosa.
- Analise o grupo da turma e seleccione crianças mais maduras para agirem como “amigos” ou para formarem um “círculo de amigos”.
O sistema de “amigo” envolve a identificação de outra criança que esteja disposta a ajudar a criança com síndroma de Asperger a negociar as dificuldades. É possível formar “círculos de amigos” para que o ónus não recaia apenas sobre uma criança.
Desenvolvimento das capacidades e da compreensão da criança
O sentido de si próprio. Diz-se frequentemente que as crianças com síndroma de Asperger têm um elevado nível de egocentrismo. Dito desta maneira, parece que elas optam por agir assim – mas isso não é verdade! Muitas vezes, nem sequer compreendem os seus sentimentos e comportamentos. Pode acontecer a criança preocupar-se com o seu próprio bem-estar. As perguntas de adolescentes capazes como, por exemplo: “Estarei louco?” causam preocupação em todos os que os rodeiam. O objectivo da intervenção consiste em aumentar a confiança da criança em si própria enquanto indivíduo, uma vez que uma maior autoconfiança reduz a ansiedade. Devem ser tidos em conta os seguintes aspectos:
- É necessário esforçarmo-nos por fazer com que a criança tenha uma imagem positiva de si própria.
- Chegará o momento certo para comunicar à criança os antecedentes das suas dificuldades. Isto terá de ser debatido entre os pais e os profissionais.
- Depois de a criança estar ciente do “diagnóstico”, desencoraje-a de culpar a síndroma de Asperger por tudo e por nada. Encoraje-a a pensar em estratégias alternativas.
- Poderá ser necessário ensinar a criança a desenvolver um “sentido de si própria”. Encoraje-a a reflectir no seu próprio papel nos acontecimentos e nas actividades, utilizando fotografias ou vídeos e relatos subjectivos.
Interacção com terceiros. As crianças com síndroma de Asperger não estão cientes das necessidades de terceiros relativamente a elas. A intervenção visa aumentar o desejo da criança para interagir com terceiros. Pode começar por aumentar a consciência da criança relativamente ao comportamento de terceiros e, em seguida, trabalhar para ensinar a criança a interagir com terceiros. Para isso, é necessário ensinar-lhe capacidades sociais específicas, de preferência em ambientes funcionais e realistas:
- Identifique as áreas de interacção social em que a criança se debate particularmente. Analise as capacidades necessárias para melhorar o seu desempenho e ensine-lhas em pequenos passos realizáveis. Por exemplo: como entrar num grupo; como falar sobre coisas interessantes para os outros – e não só sobre os seus interesses relativamente a “animais domésticos”; como se manter envolvido no assunto do grupo.
- Tire o máximo partido de actividades que se prestem a trabalhar em equipa como, por exemplo, leitura em parceria.
- Dê oportunidade à criança para jogar jogos de tabuleiro simples como, por exemplo, damas, com um parceiro. Inicialmente, o parceiro pode ser um adulto em quem ela confie, introduzindo outra criança no jogo posteriormente.
- Comece por envolver a criança em jogos de equipa simples – na sala de aula, na aula de ginástica ou no recreio. Explique muito bem o papel da criança no jogo.
Ambiente curricular
A maioria das crianças com síndroma de Asperger frequenta estabelecimentos de ensino regular. Alguns professores sentem-se sobrecarregados com a responsabilidade de ensinar uma criança dessas numa turma de 28 alunos ou mais. Não é por nenhuma razão hostil ou negativa, mas simplesmente devido a ansiedade face ao desconhecido. Pensam que podem ter de utilizar estratégias fora do repertório habitual de ensino para satisfazer as necessidades das crianças. Na realidade, o que é necessário é uma combinação de compreensão da síndroma de Asperger e de algumas práticas recomendadas na sala de aula. Os professores têm de estar informados, ser tolerantes e estabelecer empatia numa situação em que todos os funcionários estejam cientes das implicações pedagógicas desta síndroma. As limitações específicas fazem parte de tal síndroma, mas haverá muitas capacidades que a escola pode desenvolver na criança. As dificuldades da criança com esta síndroma derivam das limitações da “Teoria da Mente”, Coerência Central e Função Executiva. Deste modo, estas crianças têm uma perspectiva diferente do mundo, o que se reflecte no seu estilo de aprendizagem. O estilo de aprendizagem destas crianças é definido pelas seguintes características:
- Motivação. A criança não tem motivos competitivos. É desprovida de orgulho e vergonha, e não tem desejo de “sobressair”.
- Imitação. Embora consiga copiar o que os outros fazem, é-lhe difícil ajustar estes movimentos copiados ao seu próprio modelo de referência.
- Percepção. Existe a possibilidade de respostas incoerentes ou inesperadas aos estímulos sensoriais.
- Atenção. O foco de atenção da criança é frequentemente reduzido e/ou obsessivo. As características dos estímulos podem ser combinadas de modo idiossincrático.
- Memória. Provavelmente, a memória da criança é episódica, isto é, os acontecimentos não são arquivados no contexto em que ocorrem. É deste modo que são arquivadas listas de factos, sem nenhuma estrutura de significado que as relacione.
- Sequenciação. A criança terá dificuldade em seguir sequências. Talvez consiga estabelecer correspondência com uma sequência e, no entanto, não consegue ir além do modelo para deduzir a regra ou o princípio em que se baseia. Por esse motivo, qualquer alteração nas sequências de acontecimentos deixa a criança angustiada por não reconhecer o princípio orientador.
- Resolução de problemas. A criança tenta aprender respostas definidas para situações concretas. Pode aprender um conjunto de estratégias, mas não está ciente desse conhecimento, pelo que não é capaz de seleccionar uma estratégia adequada para uma nova situação.
Ao percorrer o Currículo Nacional, existem temas comuns que pode ser necessário diferenciar para acomodar o estilo de aprendizagem particular da criança com síndroma de Asperger. Para o ilustrar, serão de seguida analisados, do ponto de vista de tal criança, alguns elementos seleccionados do Currículo Nacional, destacando-se exemplos de possíveis dificuldades, bem como as estratégias de intervenção sugeridas.
Área Curricular: Português
Exemplo de dificuldades
A capacidade da criança para pensar com imaginação é limitada, originando problemas de escrita criativa.
Estratégias sugeridas
- Dê à criança oportunidades adicionais para escrever sobre experiências verídicas.
- Utilize estes relatos como base para desenvolver a escrita criativa.
- Por exemplo: “Foi isto que aconteceu realmente quando foste passear para a praia; mas o que aconteceria se tivesse chovido/tivesses perdido a semanada?”
- Ajude a criança, discutindo com ela as possibilidades. Limite os elementos criativos à experiência da criança.
Exemplo de dificuldades
- A criança pode ter inúmeras capacidades para a leitura.
- Consegue descodificar as palavras, mas não compreende exactamente aquilo que leu.
Estrtégias sugeridas
- Aumente a compreensão da criança, chamando-lhe a atenção para as ilustrações – “O que está acontecer nesta imagem?”
- Peça-lhe para prever o que vai acontecer em seguida. Por exemplo: “O que é que o menino vai fazer agora?”
- Peça-lhe para contar o que acaba de ler.
- Dê preferência a livros mais realistas do que fantasiosos.
- Dê à criança livre acesso a livros que não sejam de ficção. É-lhe mais fácil obter informação a partir destes livros do que de histórias.
Área Curricular: Educação Visual e Tecnológica (EVT)
Exemplo de dificuldades
O aluno tem dificuldade nos aspectos de EVT que exigem um nível significativo de pensamento criativo.
Estratégias sugeridas
Escolha um projecto que seja prático, relevante para o aluno e em que ele possa utilizar uma experiência directa recente.
Exemplo de dificuldades
Para o aluno, é difícil escolher os materiais e o equipamento a utilizar.
Estratégias sugeridas
Não sobrecarregue o aluno com demasiados itens à escolha. Não lhe apresente mais de 2 alternativas de cada vez.
Exemplo de dificuldades
O aluno distrai-se com pormenores irrelevantes. Foge completamente ao assunto e não consegue concluir a tarefa.
Estratégias sugeridas
Minimize as distracções com a disposição dos materiais. Ajude o aluno a manter-se concentrado fornecendo-lhe uma descrição escrita, com a tarefa delineada em passos explícitos.
Exemplo de dificuldades
O currículo espera que o aluno tenha uma abordagem aberta ao desenvolver as ideias e que explore um leque de soluções possíveis antes de seleccionar uma. Este aluno talvez só veja uma possibilidade, mantendo-se estritamente fiel a ela.
Estratégias sugeridas
Comece
por acompanhar a sua limitação e concentre-se em desenvolver as suas
capacidades para a descrição, o registo, a organização e o planeamento.
Posteriormente, encoraje-a a considerar outras opções e estratégias. A
utilização de fichas estruturadas pode ser extremamente útil.
Área Curricular: Matemática
Exemplo de dificuldades
A criança tem dificuldade em compreender instruções complexas.
Área Curricular: Matemática
Exemplo de dificuldades
A criança tem dificuldade em compreender instruções complexas.
Estratégias sugeridas
Simplifique a linguagem utilizada. Dê uma instrução de cada vez. Utilize objectos e imagens para ajudar a criança a compreender.
Exemplo de dificuldades
A criança tem um fascínio por números e faz incessantemente as mesmas perguntas – interrompendo a aula.
Estratégias sugeridas
Defina uma regra explícita. Diga à criança que só pode fazer a mesma pergunta, por exemplo, 3 vezes. Tente reservar algum tempo para que a criança possa colocar as suas dúvidas.
Exemplo de dificuldades
A criança tem dificuldade em compreender a linguagem Matemática. Fica particularmente confusa quando são utilizadas palavras (enunciados) diferentes com o mesmo significado. Por exemplo, multiplicar/vezes.
Estratégias sugeridas
Utilize exemplos práticos para ajudar a que as palavras façam sentido. Reúna um conjunto de palavras com conceitos relacionados, que a criança possa utilizar como referência.
Exemplo de dificuldades
A criança não tem a certeza de como responder a perguntas do tipo: “Porquê?”
Estratégias sugeridas
Sempre que possível, transforme as perguntas em afirmações, com um espaço em branco para a resposta da criança.
Área Curricular: Ciências
Exemplo de dificuldades
O aluno prefere trabalhar sozinho e resiste a ter de partilhar uma tarefa com outro aluno. As actividades de grupo apresentam ainda mais dificuldades.
Estratégias sugeridas
Seleccione cuidadosamente os parceiros e os membros dos grupos. Explique bem o papel de cada parceiro/membro do grupo. Inicialmente, talvez o aluno precise de ter um papel passivo como, por exemplo, registar os resultados. Gradualmente, prepare-o para ter um papel mais activo.
Exemplo de dificuldades
O aluno não consegue mostrar consideração pela sua própria segurança nem pela dos outros.
Estratégias sugeridas
O aluno não consegue mostrar consideração pela sua própria segurança nem pela dos outros.
É importante descrever da forma mais explícita as possíveis consequências das diferentes acções. É preciso ensiná-los a verificarem os procedimentos e incutir-lhes essa rotina.
Exemplo de dificuldades
O aluno é incapaz de pedir ajuda nas aulas.
Estratégias sugeridas
Primeiro, ensine-o a reconhecer que está “encravado”. Uma vez que ele não consegue “pôr-se no lugar” do professor, talvez não se dê conta de que este dispõe das informações de que ele precisa. Ensine-lhe especificamente como pedir a ajuda de que necessita.
Para muitas crianças com síndroma de Asperger, não é o currículo que apresenta mais dificuldades, mas sim as áreas não curriculares (p. exp.: o recreio, o intervalo para o almoço), que lhes colocam mais desafios e com que têm maior dificuldade em lidar. É o seu comportamento nestas alturas que pode começar por suscitar preocupações nos professores que podem julgar que o seu papel principal é ensinar o programa (currículo) e sentir-se menos seguros na sua capacidade para desenvolver as capacidades interpessoais da criança na medida necessária para uma criança com tal síndroma. As seguintes informações visam apresentar pontos de partida de intervenção, proporcionando exemplos de práticas recomendadas numa série de situações diferentes.
Recreio
Para a maioria das crianças, o recreio é a melhor parte do dia na escola, mas as crianças/jovens com síndroma de Asperger podem recear a hora do recreio. O recreio não é estruturado. As crianças são ruidosas e exuberantes. Não há regras. Formam-se pares e grupos. Os professores relatam frequentemente que a criança com esta síndroma não tem amigos e se comporta como um solitário no recreio. O recreio apresenta oportunidades para desenvolver as capacidades sociais da criança – desde que aceite a necessidade da criança para relaxar à sua maneira. As estratégias para ajudar a criança/jovem com este síndroma a lidar com o recreio incluem:
- Definir um “espírito” de recreio, em que se encoraja a colaboração.
- Aceitar que a criança com tal síndroma pode precisar de um tempo só para si durante o intervalo – como se fosse uma pausa das exigências sociais da sala de aula.
- Organizar jogos sociais simples e estruturados – em que o papel de cada indivíduo é óbvio. - Convidar a criança a participar.
- Encorajar a criança a observar as actividades que decorrem no recreio. Explicar essas tarefas enquanto decorrem. Arranjar uma actividade que seja apelativa para a criança e, com o apoio dos colegas, encorajá-la a participar.
- Ensinar à criança “frases introdutórias” que a ajudem a iniciar uma conversa. Muitas vezes, estas crianças/jovens até querem participar nos acontecimentos – só não sabem como fazê-lo.
Movimentar-se pela escola
No 1.º ciclo há muito movimento e espaço para muita confusão em determinadas alturas do dia. Este movimento e esta confusão aumentam significativamente nos 2.º e 3.º Ciclos e Secundário, em que existe frequentemente um movimento colectivo (do tipo “manada”) no fim de cada aula. Estas alturas podem ser de grande tensão para a criança com síndroma de Asperger, simplesmente devido ao elevado número de crianças que se deslocam numa pequena área. Além disso, os 2.º e 3.º Ciclos e Secundário acarretam outra fonte de ansiedade com a necessidade de encontrar a sala certa num edifício enorme. As estratégias para ajudar a criança com esta síndroma a lidar com estas movimentações pela escola incluem:
- Inicialmente, desfasar a chegada/partida da criança para que o volume de “tráfego” seja menos arrasador.
- Preparar as visitas às escolas dos 2.º e 3.º Ciclos e Secundário antes da transferência, para dar uma oportunidade à criança para aprender a respectiva planta.
- Estabelecer uma rede de “amigos” ou um “círculo de amigos” que estejam disponíveis para servir de “guia”.
Na cantina
As cantinas podem ser espaços muito barulhentos e muito exigentes do ponto de vista social. Espera-se que as crianças comam em público ao lado de outras seis ou oito crianças. Na maioria das escolas, a hora do almoço envolve estar na fila – uma actividade que muitas crianças com síndroma de Asperger têm dificuldade em tolerar. As estratégias para ajudar a criança/jovem a lidar com a cantina da escola incluem:
- Estabelecer regras claras, reforçadas por auxiliares visuais.
- Simular as rotinas do almoço no refeitório vazio e sossegado.
- Considerar permitir que a criança passe para o princípio ou fim da fila, em vez de ter de ficar no meio de toda a gente. Deste modo, a criança sente-se menos ameaçada.
- Alertar os auxiliares do refeitório para as dificuldades da criança e as estratégias que estão a ser utilizadas.
- Ensinar à criança capacidades de conversação simples para o ajudar a integrar-se com as outras crianças à mesa.
Pedir ajuda e resolver problemas
“Stôra! Como é que eu faço isto?” A maioria das crianças pede ajuda prontamente quando não tem a certeza de alguma coisa. As crianças com síndroma de Asperger têm muita dificuldade em fazê-lo – por uma série de motivos:
- As crianças/jovens com tal síndroma têm dificuldade em resolver problemas – em combinar os elementos de uma tarefa para as ajudarem a chegar a uma conclusão. Pedir ajuda é um dos elementos do processo de resolução de problemas. Não se apercebem necessariamente da relação entre o problema e a ajuda externa.
- Devido à capacidade limitada que estas crianças/jovens têm para reconhecer a existência de diferentes pontos de vista, podem não se aperceber de que outra pessoa possa ter uma solução para o problema com que se deparam.
Infelizmente, este comportamento pode ser confundido com preguiça ou falta de motivação – se pensarmos que a criança/jovem não quer trabalhar, e não que simplesmente não consegue avançar. As estratégias para ajudar estas crianças/jovens a pedir ajuda incluem:
- Ter consciência das tarefas em que a criança tem mais dificuldades.
- Se a criança tiver dificuldade em interpretar uma tarefa, experimente trabalhar com ela (sentado ao lado dela). Execute primeiro a tarefa e chame a atenção da criança para aquilo que está a fazer. Deixe-a experimentar fazer uma parte da tarefa. Reduza gradualmente a sua participação até a criança já conseguir trabalhar sozinha.
- Quando a criança tiver concluído a tarefa, faça-a reflectir naquilo que aprendeu, explicando-lhe aquilo que fez e como o fez.
- Ensine-a especificamente a reconhecer quando está “encravada” e como pode pedir ajuda.
Trabalhar com outros
Os professores esperam que as crianças trabalhem em grupo com outras crianças várias vezes durante o dia. Podem não se dar conta da tensão que isto pode causar numa criança com síndroma de Asperger, que tem tanta dificuldade em relacionar-se com os outros. Esta criança/jovem tem pouca ou nenhuma consciência dos sentimentos dos outros ou do impacto do seu próprio comportamento nos outros. Pode aceitar passivamente a presença de outras crianças, mas algumas crianças ficam tensas simplesmente porque estão sentadas muito perto de alguém. O contacto é unilateral – da outra criança para com ela, mas raramente é recíproco. Além disso, na escola espera-se que as crianças trabalhem em grupo (com um parceiro ou com um pequeno grupo). Com algum planeamento e preparação cuidadosos, a criança com síndroma de Asperger pode ser incluída nestas actividades. As estratégias para ajudar estas crianças/jovens a trabalhar com outros incluem:
- Ter consciência do nível de contacto social que a criança consegue tolerar sem ficar ansiosa.
- Inicialmente, permitir que a criança se sente “na ponta” durante uma actividade de grupo, possivelmente com um professor de educação especial entre ela e a criança do lado.
- Considerar dispor os lugares de uma determinada maneira. A criança pode sentir-se mais à vontade se a criança “do lado” se sentar na diagonal à frente dela – e não ao lado.
- Quando a criança já tolerar que outras crianças se sentem ao seu lado, o professor pode preparar tarefas simples em que tenham de revezar-se – inicialmente só com ela; mais tarde, com outra criança; por último, o professor retira-se, encorajando a criança a trabalhar com o parceiro.
- É possível que surjam oportunidades para utilizar as próprias capacidades ou interesses especiais da criança, talvez conseguindo que ela mostre a um parceiro como fazer alguma coisa no computador.
- Definir explicitamente e ensinar o papel de cada um numa tarefa de grupo ou numa actividade colectiva – acabando com a incerteza para a criança com tal síndroma.
A vida escolar é mais simples para as crianças com síndroma de Asperger quando os adultos que as rodeiam reconhecem até que ponto as exigências sociais resultam em tensão. A intervenção e o apoio nesta área dão frutos em termos de percepção das regras sociais aplicáveis ao ensino, dado que a tensão obsta à aprendizagem.
Em resumo:
A síndroma de Asperger é uma condição que se pensa estar enquadrada no espectro do autismo – com traços distintivos suficientes para garantir um “rótulo” próprio.
- Foi descrita pela primeira vez em 1944 pelo médico austríaco Hans Asperger, cujo trabalho foi publicado pela primeira vez em inglês em 1991.
- Caracteriza-se por limitações subtis nas três áreas de desenvolvimento: comunicação social, interacção social e imaginação social. Em certos casos, também se registam problemas adicionais de organização e coordenação motora.
- Afecta pessoas de inteligência média e acima da média.
- Pensa-se que a prevalência se situe na ordem de 36 por 10.000.
- A probabilidade de incidência é maior nos rapazes do que nas raparigas, com uma taxa de probabilidade de 10 rapazes para 1 rapariga.
- Hans Asperger considerou que a “formação” pedagógica ajudaria a criança/jovem e, para a maioria das crianças/jovens, provou-se ser verdade. Para ser eficaz, a intervenção para as crianças/jovens com síndroma de Asperger tem de assentar na compreensão da natureza de condição e das limitações fundamentais inerentes. É necessário fazer concessões em função da individualidade, uma vez que cada criança/jovem é afectada de maneira diferente.
Finalmente:
- Comece no nível da criança/jovem.
- Tente ver o mundo do ponto de vista da criança/jovem.
- Adapte o ambiente escolar de modo a facilitar a aprendizagem por parte da criança/jovem.
- Consulte os Pais e o professor de educação especial e colabore com eles.
- Considere intervenções específicas para desenvolver as capacidades da criança/jovem nas áreas de Interacção Social, Comunicação Social e Pensamento Flexível.
- Familiarize-se com as explicações psicológicas subjacentes ao estilo de aprendizagem da criança/jovem.

Bibliografia:
Attwood, T. (2006). O Síndrome de Asperger: Um guia para pais e profissionais. Lisboa: Verbo Editora.
Tucker, E. (S/d). Síndrome Asperger: Guia para professores. Associação Portuguesa do Síndrome de Asperger.
Disponível em: http://www.apsa.org.pt/
Cumine, V.; Leach, J.; Stevenson, G. (2006). Compreender a Síndroma de Asperger: Guia prático para educadores. Porto: Porto Editora
Kirby, B.L. (S/d). Asperger's Syndrome Guide For Teachers. Disponível em: http://www.udel.edu/bkirby/asperger/
Síndrome de Lowe
Síndrome de Lowe
ENQUADRAMENTO TEÓRICO
2.1 – Conceito da SL
A
SL é uma doença genética rara, também conhecida por síndrome
óculo-cerebro-renal, que provoca deficiências físicas e mentais, bem
como problemas de saúde. É uma doença de herança recessiva ligada ao
cromossoma X, caracterizada por acidose tubular renal, catarata
congénita bilateral, glaucoma, atraso mental e hipotonia muscular.
Esta
síndrome foi descoberta em 1951 pelo DR. Charles Lowe e a sua equipa e
foi apresentada em 1952 por Lowe, Terrey e Mac Lachlan (cit. por
Bresolin et al, 1999) à qual caracterizavam pela tríade: catarata
congénita bilateral, nefropatia tubular e atraso mental.
2.2 – Etiologia da SL
A SL é causada pelo mau funcionamento de um gene do qual resulta o
défice de uma enzima chamada phosphatidylinositol 4,5 – biphosphase.
Esta enzima é essencial a funcionamento metabólico de uma parte da
célula chama aparelho de Golgi.
As
funções celulares reguladas pelo aparelho de Golgi não se fazem
normalmente, o que conduz a problemas ao nível ocular (cataratas), do
fígado e do cérebro. Contudo ainda não se compreende o modo como esta
insuficiência provoca os sintomas da doença.
Esta doença hereditária é transmitida pela mãe e somente os rapazes a desenvolvem. As raparigas são apenas portadoras do gene.
2.3 – Características da SL
A
gravidade dos sinais e sintomas varia de indivíduo para indivíduo.
Geralmente os indivíduos que são afectados por esta síndrome são
afectuosos e sociáveis, gostam de música e têm um “grande” sentido de
humor.
Esta
não tem cura mas a maior parte dos sintomas pode ser atenuados através
de medicação, cirurgias e métodos de educação especial.
A esperança de vida varia entre os 30 e os 40 anos de idade.
São apresentadas no Quadro 1 alguns das possíveis características que podem ser observadas num indivíduo com SL.
Características dos indivíduos com SL
Quadro 1 – Quadro de características das crianças de SL
2.4 - Diagnóstico da SL
Segundo Ferreira (2004) existem dois tipos de diagnóstico, o etiológico e o descritivo.
De acordo com o mesmo autor, o diagnóstico etiológico “pode implicar ou
não o recurso a vários tipos de avaliações ou exames complementares”
(2004: 704), no que refere ao diagnóstico descritivo, caracteriza – se
pela referência aos vários aspectos do quadro clínico.
Assim
sendo, os pais quando sabem que são portadores do gene deficitário, ou
após o nascimento da criança, deverão recorrer aos serviços médicos para
ser efectuado o diagnóstico clínico e ser posteriormente ser elaborado o
diagnóstico etiológico, para se iniciar a elaboração de um plano de
ajudas.
Quando
não se recorre a esta estratégia de actuação, os pais desconhecem que
são portadores do gene deficitário, ou outras situações, só se consegue
detectar a SL nas crianças quando estas têm por volta de 1 ano de idade.
Assim
sendo, as famílias que apresentem casos de SL são aconselhadas a
consultar um geneticista para que se possa estudar todas as
possibilidades de risco. Para estas famílias existem vários tipos de
exame, de acordo com a situação de cada uma.
Se a mãe é portadora ou o pai tem SL existe o exame ocular, que poderá confirmar ou não se o feto está afectado.
Para além dos exames a efectuar podemos ainda constatar os caminhos a percorrer de acordo com as respostas obtidas.
São estes os exames que permitem prevenir o nascimento de crianças com SL.
No
caso de a criança estar afectada os pais poderão recorrer, ainda ao
diagnóstico de DNA. Este permite saber se a criança será apenas
portadora ou então possuidora da SL, mesmo antes de se saber o sexo da
criança.
Percorridas
todas estas etapas as famílias deverão são aconselhadas a consultar um
especialista na área dos genes afectados para estudar todas as
possibilidades, caso a gravidez esteja num nível avançado, ou então,
decidir se leva ou não a gravidez a bom termo.
2.5 - Intervenção pedagógica
Actualmente,
quando chegam à escola, “muitos alunos com NEE vêm já “sinalizados” e
são frequentemente acompanhados de “planos de recuperação”, tentados em
anos anteriores ou propostos para os meses imediatos (Correia, 1999:
109).
Por
conseguinte uma criança ou jovem com SL ao chegar à escola deverá ter
um atendimento especializado, no sentido de diagnosticar as suas
necessidades e possibilidades.
Este
diagnóstico deverá ser elaborado com base na análise atenta dos planos
e/ou outros relatórios que acompanhem o processo individual do aluno,
uma vez que estes “fornecem ao professor elementos preciosos acerca das
capacidades e das realizações do aluno, bem como elementos relativos aos
seu funcionamento no contexto da aula” (Correia, 1999: 109).
A
partir deste diagnóstico irá ser desenvolvido o seu projecto pessoal,
no qual estarão presentes os modelos de atendimento à diversidade
adequados ao aluno em questão.
Segundo
Correia, este diagnóstico deverá atender a quatro fases distintas mas
complementares: conhecimento, planificação, intervenção (preliminar,
compreensiva e transaccional) e reavaliação e deverá ser elaborado por
uma equipa multidisciplinar.
Devemos ter em consideração que, tal como refere Correia (1999: 87), “a avaliação preliminar constitui
uma das etapas mais importantes de todo o processo de avaliação para a
criança em risco educacional ou com possíveis NEE”.
No
decorrer deste processo pode ser constatado que, na escola do ensino
regular, não é possível suprimir ou minorar os problemas da criança.
Assim
sendo, esta deve ser encaminhada para os serviços de Educação Especial a
fim de se lhe possibilitar uma aprendizagem mais individualizada e
acompanhada garantindo, assim, que lhe seja facultado um bom
desenvolvimento global. Esta oportunidade, no entanto, não deve descorar
a possibilidade de cooperação entre professores do ensino regular,
professores de educação especial, pais e outros técnicos que
eventualmente sejam consultados.
Nos
serviços de Educação Especial, a criança / jovem terá o apoio
necessário dos diversos técnicos a fim de elevarem as suas capacidades,
designadamente: Professor / Educador; Auxiliares de Educação;
Psicomotricista; Terapeuta da Fala; Terapeuta Ocupacional; Psicólogo;
entre outros.
Deve
existir, por parte dos técnicos, uma preocupação acérrima em trabalhar
as áreas Psicomotora, Cognitiva, Perceptiva-Motora, Sócio-afectiva e
Autonomia pessoal, social.
A
criança / jovem com SL deve ser estimulada a todos os níveis devendo
beneficiar de actividades diversificadas no âmbito da Psicomotricidade,
da Terapia Ocupacional, da Fisioterapia, da Terapia da Fala, de Apoio
Psicológico, da Estimulação Global (ou estimulação académica numa fase
inicial), da Expressões Musical e Dramática, da Natação ou Actividades
Aquáticas, da Autonomia Pessoal e Social, a fim de abordar e desenvolver
ao máximo a sua autonomia pessoal no que concerne à higiene,
alimentação, segurança, entre outras.
O envolvimento e a comunicação entre os técnicos são de extrema importância para um desenvolvimento mais sustentado.
Para
além disto, é necessário que o aluno com SL disponha de recursos
materiais e espaciais adequados ao seu desenvolvimento. Nomeadamente,
materiais lúdicos e pedagógicos e espaços amplos devidamente
apetrechados.
É
importante que estes alunos contactem com outras pessoas (sem ser
colegas, técnicos e encarregados de educação), assim como espaços
exteriores (ao ar livre).
Todos
os materiais devem estimular o aluno para o seu desenvolvimento físico e
mental trabalhando sempre estímulos como: a luz, a cor, o som, a
textura.
Nesta fase é importante que a criança / jovem seja estimulada ao máximo para que se verifiquem evoluções significativas.
3- PROCEDIMENTOS METODOLÓGICOS
A
investigação realizada para o presente trabalho baseia-se no estudo de
um caso real. Foi escolhida esta metodologia pelo facto de se considerar
a mais adequado face ao assunto em estudo.
O
estudo de caso tem como objectivo, tal como refere Mucchielli (1968), o
estudo de situações dialécticas ou organizativas que podem ser objecto
de análise e reflexão e que conduzem à descoberta de relações
significativas entre diversos factos, permitindo uma interpretação
contextualizada do investigador.
Por
seu turno, Shulman (1989), defende que o estudo de caso ajuda o
investigador a interpretar a realidade estudada, proporcionando-lhe
oportunidade para uma reflexão sobre as experiências dos outros e
constituindo uma poderosa ferramenta de investigação.
O
estudo de caso é, de facto, uma das estratégias privilegiadas pelos
estudos qualitativos, permitindo explorar a subjectividade dos fenómenos
educativos, com vista a “estabelecer generalizações acerca da mais
ampla população à qual pertence a unidade em estudo” (Cohen e Manion,
1990:164).
Este
tipo de estudo permite, segundo Poisson (1990), percepcionar os
processos mais que os resultados, bem como o modo como os participantes
interpretam as suas experiências e como lhes dão significado.
Podemos
considerar que se trata de um trabalho empírico que se baseia
essencialmente no trabalho de campo e que ao estudar uma entidade, neste
caso professores e alunos na sua pratica, tenta tirar partido de
instrumentos como a observação naturalista e participante e a
documentação existente (Yin, 1989).
A
estratégia associada ao estudo de caso permite que numa atitude de
introspecção seja facilitada a compreensão das relações que se
estabelecem durante o processo de trabalho.
O
estudo de caso apresenta algumas características que importa referir:
tem por objectivo um fenómeno contemporâneo da vida real; as fronteiras
entre o fenómeno estudado e o contexto são nitidamente demarcadas; o
investigador utiliza fontes múltiplas de dados (Yin 1989).
Dentro
do estudo de caso utilizamos, para a recolha de dados, a análise
documental dos relatórios médicos clínicos e psicológicos, os PEI’s e as
Programações elaboradas para o Roberto e ainda nos socorremos de
informações de carácter informal, recolhidas por observação directa e
conversas informais.
Prof. Fernanda Ferreira
Prof. Marilia Dias
Prof. Pedro Santos
**************************************************************************************************************************************************
Síndrome de West
Síndrome de West
O
Síndrome de West é um tipo raro de epilepsia, chamada de "epilepsia
mioclónica". As convulsões que a doença apresenta são chamadas de
mioclonias e podem ser de flexão ou de extensão, e afectam geralmente
crianças com menos de um ano de idade. São como se, de repente, a
criança se assustasse e quisesse agarrar uma bola sobre o seu corpo.
Os
espasmos são diferentes para cada criança. Podem ser tão leves no
início que não são notados ou pode-se pensar que originam-se de cólicas.
Cada espasmo começa repentinamente e dura menos de alguns segundos.
Tipicamente, os braços se distendem e a cabeça pode pender para a frente
e os olhos fixam-se em um ponto acima (Tic de SAALAM).
No
início, a criança pode experimentar somente um ou dois espasmos por
vez, mas, no decorrer de um período de dias ou semanas, estes evoluem
para dúzias de espasmos que ocorrem em intervalos de poucos segundos. As
convulsões são de difícil controlo, e a criança pode chegar a ter mais
de 100 convulsões por dia.
Cada
espasmo é uma crise epiléptica (ataque epiléptico) composta de uma
série de movimentos descontrolados, causados por um excesso de
actividade eléctrica no cérebro.
Um
eletroencefalograma (EEG) pode registrar esta actividade de forma
segura e indolor. Um EEG em um bebé com espasmos infantis não apresentam
os ritmos brandos e regulares que usualmente se apresenta nessa idade.
Ao contrário, ocorrem súbitas eclosões de actividades eléctricas,
algumas de alto potencial e o registro do EEG aparenta estar caótico.
Muitas dessas alterações eléctricas tornam-se mais marcantes à medida em
que a criança adormece. Este padrão é chamado de "Hypsarritmia"
(hypsos=altura e rhytmos= ritmo).
Estes
ataques foram primeiramente descritos pelo Dr. West (1841) em relação
ao seu próprio filho. A Síndrome de West é multifactorial e certos casos
pode ter sucetibilidade poligênica ou pode ser completamente ambiental.
Algumas crianças podem chorar e/ou gritar antes ou após as convulsões e
mostram-se geralmente muito irritadas. O período mais crítico para as
convulsões são a hora de dormir ou de acordar, onde a Síndrome apresenta
toda a sua face mais cruel.
O
presente Síndrome apresenta um padrão eletroencefalográfico
característico, chamado de Hypsarritmia, é um atraso psicomotor, que
pode variar do leve ao severo, dependendo da evolução do caso. Os
espasmos infantis causados pela síndrome não possuem uma causa única.
Eles surgem como resultado de muitas e diferentes condições. Algumas
vezes ocorrem em bebés com desenvolvimento abaixo da média e em relação
aos quais já se sabe que são possuidores de condições neurológicas que
afectam o desenvolvimento do cérebro.
Tais
condições podem ter seu início antes do nascimento, por volta do tempo
que antecede o nascimento ou como resultado de doença nos primeiros
meses de vida. Alguns poucos exemplos são: Lisencefalia (um padrão
anormal de desenvolvimento do cérebro começando nos primeiros estágios
da gestação); Encefalopatia neonatal (quando os bebés mostram-se muito
doentes imediatamente após o nascimento, como resultado de um
insuficiente volume de sangue fornecido ao cérebro durante a gestação ou
durante o nascimento); Meningite (nas primeiras fases de vida). Em
outras crianças, nenhum problema neurológico será notado até que os
espasmos infantis comecem a se manifestar. Testes diagnosticarão
condições neurológicas em algumas destas crianças, entretanto, em outras
nenhuma causa será detectada, mesmo após os mais variados testes. Nas
investigações a tomografia computorizada tem detectado patologias em 75 a
90% dos casos de Síndrome de West, entretanto, a ressonância magnética
tem se mostrado mais sensitiva que a tomografia computorizada em
detectar lesões em pacientes com crises parciais.
ACTH
– É a abreviatura de Hormônio Adrenocorticotrófico ou corticotrofina,
que é um hormônio secretado pela Hipófise (glândula pituitária). O ACTH é
o responsável pela estimulação da secreção da Cortisona e compostos
relacionados pelas glândulas supra-renais. Na sua forma sintética (ou
seja, produzida por laboratórios farmacêuticos), este hormônio é usado
como medicação corticoesteróide para o tratamento da Síndrome de West e
convulsões mioclónicas na infância. E é necessário que seja
permanentemente monitorizado, durante o seu uso na forma injectável por
especialistas (neuropediatras) devido a ocorrência de efeitos colaterais
e indesejáveis, com o comprometimento de outros órgãos e sistemas além
do SNC.
CONVULSÕES
E EPILEPSIA - As convulsões são contracções súbitas e involuntárias de
músculos voluntários do corpo, que ocorrem subitamente e têm aparência
de perda de controle da postura física, estando associadas, nas
paralisias cerebrais, aos quadros epilépticos. As Epilepsias são
distúrbios intermitentes das funções do cérebro, frequentemente
associados a distúrbios da consciência. O termo é plural pois abrange um
enorme grupo de transtornos neurológicos e psiquiátricos. O tipo mais
conhecido é o chamado de "Grande Mal", caracterizado por episódios
recorrentes de convulsões generalizadas, nas quais o corpo todo
estremece numa série de curtos espasmos. Os chamados "ataques"
epilépticos variam desde os espasmos, mioclonias, ausências, convulsões
febris na infância até os acessos psicomotores em adultos. Actualmente
se classificam as convulsões epilépticas em dois grandes grupos:
Parciais e Generalizadas.
As
epilepsias são mudanças bruscas e repentinas no funcionamento
bio-elétrico do cérebro e normalmente comparadas a um "curto-circuito"
momentâneo que afectam as células nervosas (neurónios), fazendo parte de
uma disfunção do Sistema Nervoso Central. Nos "ataques" ou crises
epilépticas podem ocorrer perda de consciência momentânea, acompanhada
de distúrbios, tais como: abalos musculares, movimentos bruscos, perda
do equilíbrio corporal, alterações dos movimentos e das acções de uma
pessoa. Posteriormente, há uma pausa, um repouso devido a exaustão das
células nervosas, seguindo um período de confusão mental e/ou
sonolência. Um aspecto importante deve ser ressaltado, AS EPILEPSIAS,
ASSIM COMO AS SÍNDROMES RARAS NÃO SÃO CONTAGIOSAS, podem estar
associadas a quadros de natureza genética, sendo que 70% dos casos são
de causas desconhecidas.
Factores que podem levar a um quadro epiléptico:
-Traumas na cabeça (acidentes automobilísticos, prática de desporto violento, etc.); -Tumores cerebrais;
-Doenças
neurológicas crónicas, meningites, doenças neurológicas crónicas,
encefalites virais ou AIDS e Lesões ocorridas no período gestacional,
pré, peri ou mesmo pós-natal, que possam afectar o desenvolvimento do
cérebro do feto durante a gravidez.
Em
casos extremamente graves (quadros epilépticos parciais complexos)
existe a possibilidade de uma intervenção neurocirúrgica. Por ser um
tratamento invasivo é necessário uma rigorosa avaliação dos riscos e dos
benefícios, pois nem todas as crianças estão aptas a uma cirurgia deste
tipo.
Actualmente, existem experiências sobre a utilização de Marcapassos Cerebrais que ajudam na organização bio-eléctrica do SNC.

CORTEX
CEREBRAL - é a camada mais externa do cérebro, que contém a chamada
Substância Cinzenta, responsável pelas funções e actividades mais
elevadas do Sistema Nervoso Central (SNC). Ë ligada às actividades
cognitivas e psicológicas dos seres humanos, donde uma lesão difusa
(como ocorre em alguns casos de Paralisias Cerebrais) levará a deficits
destas actividades.
ESPASMOS
- são as contracções de nossos músculos de forma involuntária. Quando
estas contracções são, persistentes e não podem ser interrompidas pela
nossa vontade, teremos um quadro de espasticidade.
MICROCEFALIA
- Quando o crânio tem o seu desenvolvimento prejudicado, ficando o
cérebro ( em seu interior) com seu volume e tamanho diminuídos. Esta
situação pode ocorrer em diversas formas de deficiências, como as
decorrentes de lesão cerebral, do tipo paralisias cerebrais.
TÔNUS
- É um estado natural de tensão dos músculos do corpo. Mantêm-se
através de uma reflexa que conserva os músculos levemente tensos. Quando
temos a perda de tónus ( flacidez ou hipotonia) temos um distúrbio da
parte do SNC responsável por este reflexo, enquanto que um aumento
patológico do tónus (hipertonia) pode gerar a espasticidade, indicando
um dano em nível cortical do SNC, constituindo o quadro de paralisia
cerebral espástica, por exemplo.
DIETA
CETOGÊNICA - É uma dieta programada e RIGOROSAMENTE ACOMPANHADA POR
MÉDICOS que induz o organismo humano a produzir uma modificação química
chamada de cetose. É utilizada em casos de epilepsia refractária a
medicação anticonvulsivante. O tratamento tem como base uma dieta rica
em gorduras, e muito pobre em carboidratos e proteínas o que induz a
produção de corpos cetónicos. Este tratamento ainda carece de maiores
pesquisas científicas.
DELFINOTERAPIA
O
Aquário do Zoológico de Aragón (Cidade do México) vem desenvolvendo um
trabalho inovador para crianças entre 6 e 16 anos portadoras de
problemas neurológicos, como paralisias cerebrais, Síndrome de West e
Síndrome de Down.
O
tratamento consiste na interacção do ambiente (água morna, música
suave), terapeutas e golfinhos, são de seis a oito sessões diárias de 15
minutos cada, sendo que os resultados estão sendo considerados
animadores. Os golfinhos brincam e entram em contacto com a criança, o
cetáceo emite um som agudo que tem a propriedade de estimular o sistema
nervoso central, possibilitando a melhora da plasticidade cerebral,
favorecendo os sistemas psicomotor e a fala da criança. O Human Dolphin
Trerapy Center, organização americana dedicada à terapia com golfinhos,
informa que mais de um milhão de crianças afectadas por paralisias
cerebrais ou dificuldades para aprender, conseguiram melhorar o
desempenho depois de iniciar o tratamento. O tratamento é acompanhado
por um grupo de médicos, que acompanham as sessões além de desenvolverem
exercícios físicos aquáticos para as crianças. A delfinoterapia tem
propiciado inúmeras melhoras no quadro clínico dos pacientes,
entretanto, as lesões neurológicas não desaparecem.
**********************************************************************************************
Síndrome de Edwards ou Trissomia 18
 bebé com trissomia 18 Independente da idade, tem risco de ter um
defeito cromossómico em seu feto. A maioria dos pacientes com a
trissomia do cromossoma 18 apresenta trissomia regular sem mosaicismo,
isto é, cariótipo 47, XX ou XY, +18. Entre os restantes, cerca de metade
é constituído por casos de mosaicismo e outro tanto por situações mais
complexas, como aneuploidias duplas, translocações. Destes, cerca de 80%
dos casos são devidos a uma translocação envolvendo todo ou quase todo o
cromossoma 18, o qual pode ser herdado ou adquirido de novo a partir de
um progenitor transportador. Estudos recentes demonstram que, na maior
parte dos casos (85%), o erro ocorre na disjunção cromossómica da meiose
materna, e somente 15% da meiose paterna. O primeiro caso de trissomia
18 foi descrito por Edwards, no ano de 1960, daí o nome Síndrome de
Edwards.
bebé com trissomia 18 Independente da idade, tem risco de ter um
defeito cromossómico em seu feto. A maioria dos pacientes com a
trissomia do cromossoma 18 apresenta trissomia regular sem mosaicismo,
isto é, cariótipo 47, XX ou XY, +18. Entre os restantes, cerca de metade
é constituído por casos de mosaicismo e outro tanto por situações mais
complexas, como aneuploidias duplas, translocações. Destes, cerca de 80%
dos casos são devidos a uma translocação envolvendo todo ou quase todo o
cromossoma 18, o qual pode ser herdado ou adquirido de novo a partir de
um progenitor transportador. Estudos recentes demonstram que, na maior
parte dos casos (85%), o erro ocorre na disjunção cromossómica da meiose
materna, e somente 15% da meiose paterna. O primeiro caso de trissomia
18 foi descrito por Edwards, no ano de 1960, daí o nome Síndrome de
Edwards.
As
características principais da síndrome são: atraso mental, atraso do
crescimento e, por vezes, malformação grave do coração. O crânio é
excessivamente alongado na região occipital e o pavilhão das orelhas
apresenta poucos sulcos. A boca é pequena e o pescoço geralmente muito
curto. Há uma grande distância intermamilar e os genitais externos são
anómalos. O dedo indicador é maior que os outros e flexionado sobre o
dedo médio. Os pés têm as plantas arqueadas e as unhas costumam ser
hipoplásticas. Não há tratamento específico e conhecido por Síndrome de
Edward.
in http://mutacoesgeneticas.blogs.sapo.pt/4123.html
|

















Nenhum comentário:
Postar um comentário
FICAREI MUITO FELIZ SE DEIXAR UM COMENTÁRIO!